Neurofibrosarcome en dehors d’une neurofibromatose
Le neurofibrosarcome est une tumeur maligne de la gaine des nerfs périphériques. Elle est exceptionnelle dans la population générale et se voit habituellement dans le cadre de la neurofibromatose de Recklinghausen. Nous rapportons le cas d’une femme âgée de 53 ans qui a été hospitalisée pour récidive d’un panaris osteitique du pouce droit. L’examen anatomopathologique après excision a conclu à un neurofibrosarcome du pouce. Une amputation du pouce a été réalisée. Trois ans après, la patiente a présenté une tuméfaction du coude droit. L’examen anatomopathologique après exérèse a conclu à un neurofibrosarcome. Le bilan d’extension était sans anomalies et l’examen clinique et paraclinique a éliminé le diagnostic de neurofibromatose type 1. Le neurofibrosarcome est une tumeur très spécifique de la neurofibromatose de type 1 (NF1). Les localisations multiples d’emblée ne sont décrites que chez les patients atteints de NF1. La deuxième localisation pourrait être une métastase unique par voie lymphatique ou une deuxième localisation chez une patiente ayant une forme infraclinique de neurofibromatose.
A propos
Introduction
Le neurofibrosarcome est une tumeur maligne de la gaine des nerfs périphériques. On l’appelle également schwannome malin, sarcome neurogène, ou MPNST (malignant peripheral nerve sheath tumor) [1, 2]. C’est une tumeur exceptionnelle dans la population générale et se voit généralement dans le cadre de la neurofibromatose de Recklinghausen [3]. La tumeur se développe le plus souvent lors de la seconde ou de la troisième décade, mais elle semble également survenir plus tôt chez les patients atteints de neurofibromatose [4]. Nous rapportons deux neurofibrosarcomes observés à 3 ans d’intervalle chez une patiente non atteinte de neurofibromatose.
Observation
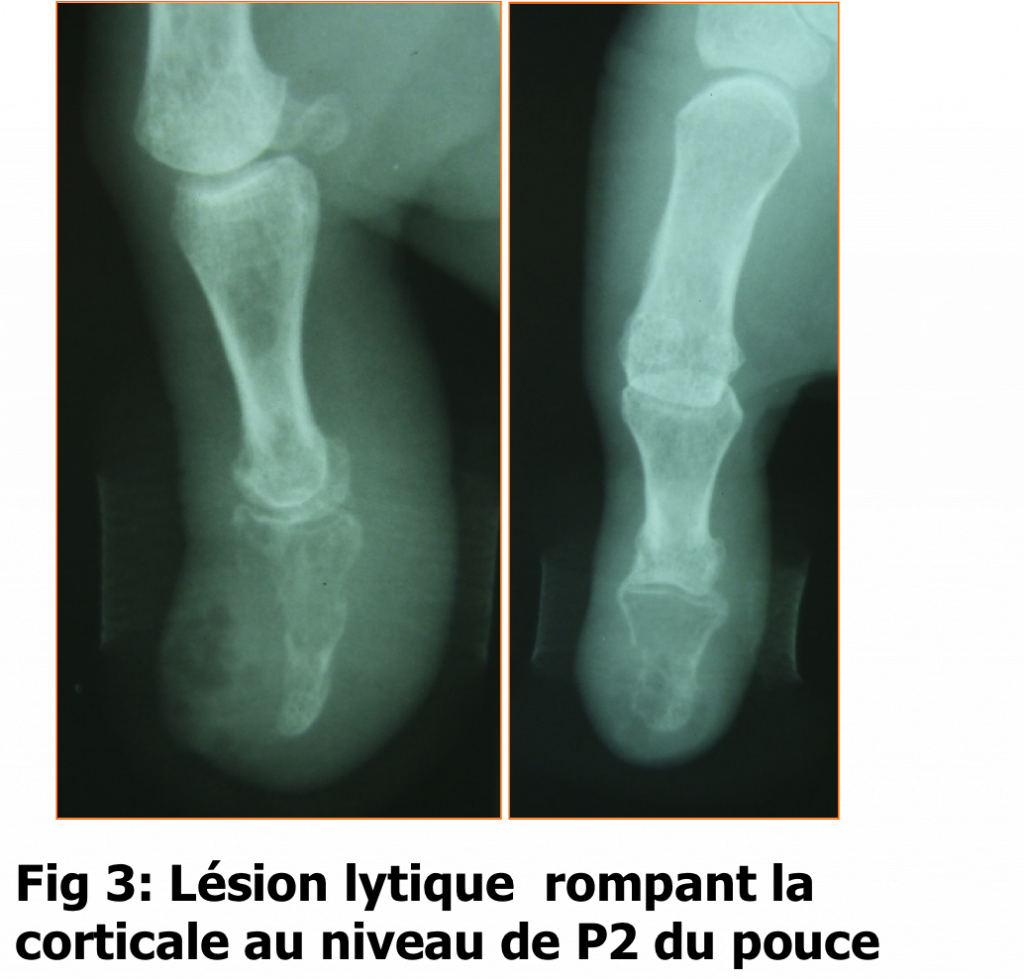
JS est une femme âgée de 53 ans qui a été hospitalisée en 2006 pour récidive d’un panaris osteitique du pouce droit, excisé 15 jours auparavant (Fig.1). La radiographie du pouce a montré une image lytique de P2 avec effraction de la corticale dorsale (Fig.2). Une excision avec examen bactériologique et anatomopathologique ont été réalisés. L’examen bactériologique n’a pas isolé de germe, mais l’examen anatomopathologique a montré la présence d’un épiderme acanthosique, par place ulcéré associé à une prolifération fuso-cellulaire, de densité faible à modérée et à noyau ondulé peu atypique. L’examen immunohistochimique a montré une positivité de la vimentine et PS100 et une négativité de D34. Cet examen a conclue à un neurofibrosarcome du pouce. Une amputation trans MP du pouce a été réalisée, sans traitement adjuvant. Le bilan d’extension était sans anomalies. Le scanner thoraco-abdominal a montré un nodule surrénalien droit et la scintigraphie osseuse a été sans anomalies.
Trois ans après, la patiente a présenté une tuméfaction dure et douloureuse mesurant 4 cm de grand axe, siégeant à la face antéro-externe du coude droit (Fig.3). L’échographie a montré une masse tissulaire sous cutanée bien limitée, extra-aponévrotique. Une biopsie exérèse, emportant la peau en regard et l’aponévrose a été faite (Fig.4). L’examen anatomopathologique a montré un nodule sous cutané bien limité par une capsule fibreuse épaisse. Les cellules tumorales étaient positives à la Vimentine et la PS100 et 5 à 10% des cellules expriment le Ki67. Cet examen a conclut à un neurofibrosarcome grade 3 OMS, complètement réséqué. Le bilan d’extension était sans anomalies et l’examen clinique et para clinique ont éliminé le diagnostic de neurofibromatose. Au recul de 2 ans, la patiente est en bon état de santé, ne présente pas de récidive tumorale et le bila d’extension n’a pas montré de métastases.
Discussion
Le neurofibrosarcome est une tumeur très spécifique de la neurofibromatose de type 1 (NF1) [3]. Le risque de développer un neurofibrosarcome dans la population générale est estimé de 0,001% ; ce risque atteint 5% chez les patients atteints de NF1 (5, 6, 7). Le neurofibrosarcome touche surtout le tronc (50%), les extrémités (30%), la région cervico-faciale (20%). Il survient dans 60% des cas au sein d’un neurofibrome plexiforme préexistant [8]. Ce dernier est alors considéré comme une tumeur précancéreuse. Un traitement antérieur par radiothérapie est parfois avancé comme facteur de risque de développer un neurofibrosarcome. Le pronostic des neurofibrosarcomes est mauvais, avec un taux de survie à 5 ans variant selon les études, de 48 à 58%. Les taux de récidive varient de 38 à 45%. On observe des métastases dans 50 à 80% des cas, localisées le plus souvent dans le poumon, les relais lymphatiques, et le foie [7].
Les localisations multiples d’emblée n’ont été décrites que chez les patients atteints de NF1 [9]. Doorns et al [10] ont rapporté une 2ème localisation primitive chez 45% des patients atteints de NF1, alors qu’ils n’ont trouvé aucune 2ème localisation primitive chez les patients sans NF1. En fait, les neurofibromes plexiformes sont susceptibles de se transformer en neurofibrosarcome à cause de la présence de cellules de Schwann peu différentiées qui semblent être susceptible d’induire une transformation maligne. La surveillance des patients atteints de NF1 et porteurs de neurofibromes plexiformes doit donc être très attentive, encore plus s’ils sont nombreux, ou étendus [11]. L’apparition de douleurs chez un patient atteint de NF1 et porteur de neurofibromes plexiformes doit faire suspecter une transformation en neurofibrosarcome. Il en est de même pour l’augmentation rapide de la taille de la tumeur, le durcissement de sa consistance ou l’apparition d’un déficit neurologique. Cependant, les neurofibromes plexiformes peuvent présenter des périodes de croissance rapide, suivie de périodes de quiescence relative et l’évolutivité n’est pas toujours synonyme de malignité (6).
Notre cas rapporté pourrait être considéré comme une localisation multiple d’un neurofibrosarcome à en dehors d’une neurofbromatose de type 1. Dans les deux cas, la tumeur n’a pas pris naissance à partir d’un tronc nerveux, mais à partir de branches nerveuses. La première localisation a montré le caractère agressif de cette tumeur avec envahissement osseux et cutané. La deuxième localisation, apparue 3 ans plus tard au niveau du coude, ne peut pas être considérée comme récidive locale car elle est située à distance de la première localisation. Elle pourrait être considérée comme métastase par voie lymphatique de la première localisation, malgré l’absence d’atteinte ganglionnaire et de métastase pulmonaire, ou une 2ème localisation primitive de neurofibrosarcome, malgré qu’on n’a pas trouvé dans la littérature de neurofibrosarcome multifocal en dehors de la neurofibromatose de Recklinghausen. Cette patiente pourrait avoir une forme cliniquement muette de neurofibromatose, ce qui nécessiterait une exploration cytogénétique.
Références
1- Gupta G, Maniker A. Malignant peripheral nerve sheath tumors. Neurosurg Focus 2007;22, 6:E12.
2- Sordillo PP, Helson L, Hajdu SI et al. Malignant schwannoma. Clinical characteristics, survival, and response to therapy. Cancer 1981: 47: 2503-9.
3- Fuchs B, Spinner RJ, Rock MG. Malignant peripheral nerve sheath tumors: an update. J Surg Orthop Adv 2005;14, 4:168-74.
4- Ferrari A, Bisogno G, Macaluso A, Casanova M, D’Angelo P, Pierani P et al. Soft-tissue sarcomas in children and adolescents with neurofibromatosis type 1. Cancer 2007;109, 7:1406-12.
5- Ducatman BS, Scheithauer BW, Piepgras DG, Reiman HM, Ilstrup DM. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer 1986;57:2006-21.
6- Ferner RE, Gutmann DH. International consensus statement on malignant peripheral nerve sheath tumors in neurofibromatosis. Cancer Res 1986; 62:1573-7.
7- Stark AM, Buhl R, Hugo HH & Mehdorn HM. Malignant peripheral nerve sheath tumours-report of 8 cases and review of the literature. Acta Neurochir (Wien) 2001; 143:357-63; discussion 363-4.
8- Zhu Y & Parada LF. The molecular and genetic basis of neurological tumours. Nat Rev Cancer 2002; 2 :616-26.
9- Molenaar WM, Ladde BE, Schraffordt Koops H, Dam-Meiring A. Two epithelioid malignant schwannomas in a patient with neurofibromatosis. Path Res Pract 1989; 184: 529-34.
10- Doorns PF, Molenaar WM, Buter J, Hoekstra HJ. Malignant peripheral nerve sheath tumors in patients with and without neurofibromatosis. Eur J Surg Oncol 1995; 21:78-82.
11- Wanebo JE, Malik JM, VandenBerg SR, Wanebo H J, Driesen N, Persing JA. Malignant peripheral nerve sheath tumors. Cancer 1993; 71: 1247-53.
Illustrations
Fig.1 : tuméfaction ulcérée de la pulpe du pouce, simulant un panaris récidivant.
Fig.2 : Lésion lytique rompant la corticale au niveau de P2 du pouce.
Fig.3 : Tuméfaction dure et douloureuse de 4 cm de grand axe de la face antéroexterne du coude droit.
Fig.4 : Pièce d’exérèse : tumeur de consistance ferme, bien limitée et de couleur blanchâtre à la coupe.

[url=https://fastpriligy.top/]how to take priligy[/url] I also run 50mg s proviron ed
70918248
References:
weight loss steroids for men (wolfonair.cloud)
70918248
References:
people on steroids before and after
70918248
References:
anabolic steriods online (Lacy)
70918248
References:
definition of anabolic steroid (Guillermo)
It Is a synthetic prescription anabolic steroid that is particularly designed to assist with weight reduction and
fat burning whereas preserving muscle mass.
Users might expertise slight changes in muscle tone and
body composition, however these changes will probably plateau shortly with out the stimulus of
training. The actuality is that Oxandrolone is on no account a substitute for hard work, however rather a device to reinforce
it. This is as a outcome of of Anavar being 5α-reduced; thus, it doesn’t aromatize,
that means estrogen ranges stay stable. It can also be appealing to athletes who don’t need additional
water weight when performing. Nonetheless, if customers want to maintain low ranges of visceral fats, clenbuterol may be a extra appropriate possibility (as many anabolic steroids can cause a bloated
look to the belly region). Ladies utilizing Winstrol usually experience a major reduction in physique fat
and an increase in lean muscle mass.
It can also be argued that HGH’s effects are more helpful for girls than males with reference to anti-aging, fats loss,
hair, skin, and nail health. However, one draw back we see with HGH for girls is bone and
tissue enlargement (present in long-term use). Females are inherently the smaller sex; thus, the fact of their ft,
arms, and skull rising larger is in all probability not desirable.
This before-and-after image is the results
of long-term HGH use (several years of injections). This feminine consumer administers HGH every single day for weight loss
and anti-aging benefits; however, the dosages utilized are unknown. The presence of Anavar is critical on this cycle, not solely from
a fat-burning perspective but in addition to prevent the physique from shifting
into a catabolic state. T3 can burn muscle tissue in addition to fats shops
when taken without anabolic brokers; thus,
operating T3 by itself is a potential disaster for a bodybuilder.
Myostatin is a myokine that suppresses myogenesis and inhibits muscle progress.
Thus, by lowering myostatin ranges, users can effectively enhance anabolism.
Ostarine will negatively affect HDL and LDL cholesterol levels, rising
the danger of atherosclerosis. Ostarine’s antagonistic effects on cardiac well being are less severe than those of different SARMs.
The cycle length for Anvarol is normally 8 weeks, adopted by a 1.5-week break.
For Clenbuterol, the beneficial dosage is often between 20-40mcg per day for newbies,
whereas advanced users can take up to 120mcg per day.
The cycle length for Clenbuterol is often 2 weeks on and 2 weeks off,
however some customers could choose to increase the cycle up to 6 weeks.
Anvarol, on the opposite hand, is mostly thought-about to be safe
and well-tolerated. Nonetheless, it could cause mild unwanted effects such as acne, hair loss, and decreased libido.
It can be necessary to note that Anvarol can work together with certain medications,
so it may be very important seek the guidance of
with your physician earlier than using it.
Anavar and Trenbolone, in comparison, are two steroids that are poles apart
by means of efficiency and side effects. Winstrol is a
steroid that it not supposed for use by women since it can lead to
virilization. Having stated that, the one way a girl can use
it is by taking it in very small portions. Nonetheless, there are
some guys who stack them collectively to be able to get as ripped and shredded as potential.
Nonetheless, Winstrol is extra powerful than Anavar and
may have lots of unwanted facet effects too.
Anavar (Oxandrolone) is a game-changing steroid that
can deliver outstanding results in only a matter of weeks.
To provide you with a better thought of what to anticipate, we’ve put collectively a timeline of Anavar outcomes, accompanied by real earlier than and
after footage. However, this stack will probably end in raised LDL ldl cholesterol and liver enzymes.
Not just this, it might additionally result in testosterone suppression due to Dianabol.
As a matter of reality, it’s rated as one
of the most powerful fats burners that you could lay your palms
on.
Greater pumps from clenbuterol will aid within the
fascia becoming extra supple, reducing such friction. This makes it
simpler for bodybuilders to eat in a calorie deficit and burn fats not directly (due to
an elevated metabolism). Clenbuterol additionally has direct fat-burning effects (similar to other beta-2 agonists) by accelerating the conversion of triglycerides to free
fatty acids. The best method to use Anavar is to begin with lower dosages,
and to increase over the course of eight weeks, where men should be beginning
with 20mg per day, and women from 2.5mg per day. When it involves dosage males usually take from 10mg-100mg, and women vary from 2.5mg-20mg.
As a result, users often discover substantial positive aspects in muscle dimension and power, permitting them to push their coaching to new
ranges. Not solely does Anavar assist construct muscle, however it additionally aids in shedding excess physique fat.
By boosting the body’s metabolic price, this steroid promotes fat burning, making it
a perfect alternative for these looking for a lean and ripped physique.
This is as a outcome of steroids are highly
effective medicine that can trigger critical unwanted effects if they aren’t used
appropriately. There are several supplements and steroids out there within the bodybuilding field which may be used to acquire wonderful outcomes.
There is no one-size-fits-all resolution for a Winstrol steroid cycle length.
Session with a physician is the best way to find out how lengthy to cycle Winstrol.
Maintaining a wholesome, balanced food regimen and incorporating common exercise into one’s routine are equally crucial in attaining sustainable success with these compounds.
Adequate rest and hydration, as nicely as correct supplementation, may help decrease any potential unwanted facet effects
and make sure that the user’s overall well being stays
a priority. This consists of beginning with lower doses, significantly for girls, and frequently monitoring one’s response to the substances.
As simply talked about anavar can produce some of the largest pumps you
might ever expertise. This is as a end result of
of increased water retention, ATP and glycogen contained in the muscle cells.
Such distinctive muscle fullness is just short-term (during a cycle),
however it’s an exciting impact for bodybuilders continuously attempting
to look greater and fuller. Some users report vital enhancements in body composition and strength, while others may expertise minimal results or adverse reactions.
ACut is manufactured in an FDA-approved lab within the US and the UK
and it mimics Anavar to have the ability to expertise
all the advantages of Anavar minus its unwanted effects.
Any questions associated to this subject must be directed towards r/testosterone.Please read the foundations and FAQ part earlier than posting.
It has a quantity of harmful unwanted effects illegal in most countries –
including the Usa. The dosage is between 20 – 40mg per day for females before experiencing
serious side effects. Men can take as much as 100mg of Anavar per
day, however this will trigger critical side effects. Additionally,
Anvarol might help to burn stored fats for gas, providing you
with a leaner physique.
As with male users, adherence to a well-designed excercise plan and a nutritious diet is important for maximizing the benefits of Anavar.
By integrating these key elements, feminine customers can experience the total
potential of this versatile substance. The
progress could be equally spectacular for women completing 4 weeks of an Anavar cycle.
The transformation may embrace a rise in lean muscle mass, improved muscle tone, and
enhanced strength. Customers could observe a more agency, sculpted look taking form among the
many notable changes. In addition to these constructive adjustments, males might expertise a discount in body fat, because the
substance aids in metabolism acceleration.
References:
good things about steriods (Koubry.Com)
Gaining vital amount of muscle mass isn’t the objective of such a
stack. Pondering you would achieve a lot of muscles with this
stack is going to depart you disappointed. There are much better options like for
example Dianabol, Anadrol or Trenbolone – they are much extra powerful coming with their
very own set of benefits and downsides. But on this cycle we’ve Anavar which, though
may be very gentle compound, even in terms of liver
hepatotoxicity, liver issues are nonetheless attainable to happen. Utilizing high ranges, you get excessive testosterone amount in your
physique, it converts into estrogen and due to this fact you’ve means too high ranges of estrogen. The high estrogen levels
is the primary cause why gynecomastia is fashioned (there’s an accumulation of breast tissue in males).
Some like to reside on the sting, whereas there are these
of us who want to balance the advantages and risks. I wouldn’t want to go beyond
six weeks if any greater than 50mg is being taken (rarely).
However that’s only one facet of the story… Efficiency
doses take things to a new degree because we wish
to profit from Anavar’s anabolic effects beyond what’s required in medical therapies.
Anavar is a fast-acting steroid derived from DHT (dihydrotestosterone)4
with a half-life of 8 to 10 hours. It has been a widely used, respected, and very popular steroid for a really lengthy time
and is considered one of the few that females can also use because of its gentle androgenic effects.
It Is imperative for users to carefully handle the cycle,
adapting their coaching and dietary strategies to maximise these benefits.
First, Anavar is a very delicate steroid when it comes
to its effects on testosterone levels. In the PED subject, Testosterone is used as a base – or as an entire cycle for newer users.
This offers users with base-level libido and well being effects, while other medicine will present strength and
progress results. Using each of these medicine can cause extra physique mass, leading to greater blood stress.
That’s why it’s important to do cardiovascular exercise,
drink extra water, scale back sodium intake, and lose excess weight.
For females, who wish to build muscle or redefine their our bodies,
the outcomes of this cycle could be astonishing.
Testosterone will increase estrogen ranges, permitting for regular male functioning and stimulating muscle growth.
Anavar is a good alternative for those trying to add
muscle mass and strength, while Check is ideal for those
trying to bulk up. When used accurately, you can expect
to see significant gains in each strength and size.
Some supplements like Omega Fatty Acids and Grape Seed Extract can decrease LDL
cholesterol levels. Anavar, a drug, might lower total levels of cholesterol and
blood stress.
Anavar is a steroid which will have unwanted facet
effects, which may concern those that need to construct
muscle and enhance physical performance. The use of Anavar, an anabolic steroid, can have several potential unfavorable consequences on one’s
health. When these two medication are combined, they work collectively to have a greater impact.
Anavar helps keep muscle mass during slicing, while Clenbuterol focuses on burning fat.
Even although Anavar has mild unwanted effects, it’s nonetheless a steroid
and no one knows how his body would react to it. It’s even attainable that your body may by no means get again to normal
hormonal manufacturing even after a PCT. Last, but not the least, Anavar is even much
less damaging to the liver as compared to many
different steroids like Deca or Dianabol.
Not solely this, but it can block Estrogen at the breast tissue as properly.
Lastly, it will promote better Cholesterol ranges through the Liver as nicely.
Earlier Than you do a PCT (and before you even cycle) you have
to be operating blood checks. These all play important roles in your well being, and to see how and when to cycle subsequent, you have to know these three things.
When working with hormone levels, you sadly can not just take aim and
hope for the best. You have to make sure about what your normal ranges are (even earlier than biking so you could have a reference point) and
you need to know what the Anabolic Steroid(s) did.
It’s crucial to find the best stability to attain optimal results whereas minimizing potential unwanted
side effects. To guarantee safety and effectiveness,
it is strongly recommended to frequently monitor hormone ranges and overall
health by way of blood checks in the course of the cycle.
Moreover, you will need to comply with post-cycle therapy
pointers to aid in natural testosterone recovery and mitigate potential side effects.
Bear In Mind, discovering the suitable dosages for Testosterone and Anavar is a personalized course of that requires cautious consideration of your particular person wants and
targets.
With Anavar, scientists attempted to formulate a compound that promoted anabolism in sufferers affected by cachexia (muscle wasting) and
delayed growth, including males, ladies, and children (4).
Thus, it was their intention for Anavar to be free from masculinizing effects.
However, our patients sometimes use Sustanon 250 in bulking cycles, the place maximum muscle gain is the aim.
We have seen notable results achieved from working Sustanon on its own, yet some bodybuilders choose to stack it with
different steroids which would possibly be also fitted
to adding mass, such as Anadrol. However, you will want to note that Anavar is a
robust drug, and it can have unwanted side effects.
These side effects can include liver injury, hypertension, and
increased levels of cholesterol. It is important to speak
to your doctor earlier than taking Anavar, especially in case you
have any underlying well being situations.
TRT is a long-term therapy and it is very important talk about the risks
and advantages together with your doctor before starting remedy.
It is a medical therapy that entails the alternative of
testosterone in men who’ve low ranges of the hormone.
When it involves optimizing efficiency and reaching fitness targets, athletes and individuals present
process Testosterone Substitute Therapy (TRT) often seek ways to reinforce their outcomes.
It’s extra compatible with female bulking as a outcome of most ladies are
not looking for the identical gains as males; simply slight enhance in muscularity.
Raise numbers beneath are at seventy eight kg, however had been ofc lower initially
due to the weightloss. To evaluate algorithm
efficiency, we carried out analyses on simulated datasets,
systematically various the variety of variables, colliders, and
time lags in symptom covariation.
Nonetheless, if you use excessive doses and long cycles, you’ll be putting your kidneys – and different organs – at great threat of harm.
Men on a chopping cycle with Anavar can count on an excellent fats loss,
and it will be quite quick because Anavar is a quick-acting steroid that you’ll only use
for eight weeks max. Using Anavar at low to moderate doses is about as secure as it could get
for anabolic steroid use. As with many different compounds, it is
unknown for its extreme unwanted effects. However abuse Anavar beyond the recommended usage patterns,
and you do set your self up for an unsafe steroid experience that
may and will injury your health.
On the opposite, Anavar, though not resulting in substantial bodily alterations,
proves its effectiveness in the best way it augments power
and prolongs endurance. Hence, the selection between the two often depends
on particular person health targets. Transitioning to
the topic of Anavar, also called Oxandrolone, we encounter one other favored choice
amongst people in search of to boost their physical
situation. Anavar stands as a distinguished anabolic steroid celebrated for its outstanding benefits.
Therefore, if Anavar is taken with the intention of bulking
and gaining lean mass, then a small calorie surplus could also be adopted to enhance muscle and energy results.
We discover Anavar peaks quick within the bloodstream because of its short half-life of
9–10 hours. In our expertise, users commonly notice an enchancment in physique composition within the first
two weeks. In the previously cited examine, fat loss was everlasting
for participants. Thus, the hormonal lipolytic results of Anavar could
also be preserved even when a person is sedentary post-Anavar
cycle. When Anavar (oxandrolone) was first produced, it was
authorized to buy for bodybuilding purposes. Medical Doctors would
prescribe it to people wanting bigger muscles, extra energy,
or to burn excess fats.
References:
Steroid Stacks And Cycles (Rosaparks-Ci.Com)
Any potential development of feet, palms, and other exterior features can be noticeable and not
likely desired by most feminine HGH customers. Treatment of HGH deficiency
is considered one of the main medical uses for artificial growth hormone.
HGH is also used to help with muscle wasting-related situations like HIV, where the patient can otherwise struggle to take care of and gain lean muscle weight.
Victims of burns are potential beneficiaries
of HGH as well due to its wonderful ability to advertise collagen synthesis.
The compounds Dianabol and Anadrol are extremely estrogenic oral
steroids which might be both extraordinarily efficacious in building muscle
and energy. Our intercourse hormone-binding globulin (SHBG) checks have indicated severe testosterone suppression from
this stack.
Anabolic steroid use amongst females for performance enhancement is unsurprisingly significantly lower than among males.
For this purpose, female steroid use is a topic where not lots of information exists and one that is not typically discussed within bodybuilding communities.
The traditional TRT dosage of Testosterone cypionate
is between one hundred and 200mg weekly. You might even run a
low dose of HCG for further help, one thing like 100iu every different day.
You can all the time run something else alongside the Test, like
Deca-Durabolin.
This is due to Anavar being metabolized by the
kidneys, thus inflicting less stress to the liver. Clenbuterol’s
fat-burning results could be attributed to its powerful results on the central nervous system (CNS).
As previously talked about, Anavar is an costly steroid due to BTG monopolizing the market and driving the worth up, costing patients $3.75–$30 per day, depending on the dose
required.
A first-time cycle (like this one) can produce roughly 20 pounds of lean mass.
We have additionally seen power increases of 30–50 lbs from customers on compound lifts.
Although it’s popular amongst males, it is much more so among girls because of a scarcity of virilization unwanted
effects. When it comes to the world of bodybuilding, it’s fairly unimaginable to get
by way of a day without listening to the word “steroids”.
There are lots of bodybuilders worldwide who are attempting to
get the most effective muscle construct with these substances.
If you are a bodybuilder who is all the time looking for for that extra edge in bodybuilding, then you’re on the proper website.
With increased purple blood cell count and increased ATP production, muscular
endurance also improves on Anavar. Our expertise and scientific research indicate that elevated liver enzymes commonly return to normal following cycle cessation (11).
He applicable dosage of Anavar varies based mostly on particular person goals and expertise with
steroids. It is essential to follow skilled advice and cling to really helpful dosages.
There are a quantity of the reason why you may need to take oral steroids after a steroid injection. For instance, if the injection was not sufficient
to relieve your symptoms, you would possibly must take oral steroids to get the
total profit. Or, if you are taking a long-term course
of steroids, you would possibly must take oral steroids to maintain up your levels.
We have detected vital shifts in LDL and HDL cholesterol levels from this duo, leading to exceedingly hypertension recordings over
180 mmHg/120 mmHg. But their chemical constructions are somewhat distinctive, with Dianabol being a by-product of testosterone and Anadrol being a derivative of dihydrotestosterone.
Therefore, trenbolone could cause notable fats
loss because it will increase stimulation of androgen receptors (AR), inducing
lipolysis. Any of these Anadrol cycles will produce exceptional features
in muscular strength and dimension. Anadrol is one of
the most powerful and toxic steroids you can take, which is
why customers must be cautious whereas biking
it. There is evidence to counsel the alternative
is true when taking Anadrol with grapefruit juice, which we’ve
seen improve its bioavailability as a outcome of fruit inhibiting CYP3A4
(5).
Nonetheless, research present that roughly 52% of males report muscle loss when taking it (20).
We have also noticed lowered muscle hypertrophy in sufferers using Finasteride.
Thus, taking this medication might counteract the anabolic results of Anavar, causing bodybuilders to
be unhappy with their results. Due to Anavar’s
mild androgenic score, it doesn’t usually produce virilization unwanted effects in girls when taken in low to moderate doses.
Anavar’s benefits are not overly powerful, at least compared to
different anabolic steroids; due to this fact, the side effects are extra tolerable for most customers.
Thus, the risk-reward ratio on Anavar is constructive for the majority
of our patients. Clenbuterol, while not a steroid, is commonly mistakenly classified as one because of its significant fat-burning properties.
It is commonly used by athletes due to its constructive effects, without any water gains.
Winstrol, like many other oral steroids, is hepatotoxic and thus causes stress to
the liver (1). LDL levels of cholesterol will almost
actually rise, with HDL ranges turning into suppressed.
Not Like another steroids, Anavar is much less likely to lead to bloating or a
puffy appearance. Whether Or Not taken orally
or sublingually, Anavar might help athletes keep a lean and dry
physique. Winstrol will trigger a spike blood pressure,
decrease testosterone production, cause stress to the liver,
and put extra strain on the joints. Winstrol can also be extra prone
to causing androgenic unwanted effects, similar to oily skin and zits
breakouts. Hair loss on the scalp may be accelerated on Winstrol
due its androgenic results. Winstrol has
a deceptively low androgenic ranking of 30 (2) which doesn’t correlate in real-life situations.
Anavar is one of the hottest oral anabolic steroids that
have ever been launched.
But to take it a step additional, elevating your Testosterone dose can result in powerful anabolic results with muscle and strength gains.
Winstrol, also known as Stanozolol, tends to be less appropriate for women due
to its sturdy tendency to bind with SHBG (Sex Hormone-Binding
Globulin), which can result in masculinization symptoms.
Nevertheless, our research signifies that it might be safely utilized
in minimal doses. Whereas Winstrol effectively builds
muscle in girls, its efficiency implies that even small quantities are impactful.
We have determined that a daily dose of 5 mg is typically safe and minimizes the risk
of virilization.
It encourages the physique to burn fat whereas defending muscle
from being burned as vitality. As lengthy as your food plan is planned and applied correctly for a cutting part, Primobolan is an extra compound in any chopping cycle with minimal
aspect impact dangers compared with different steroids. Each novices and intermediate customers can safely use Primobolan up to 150mg day by day, though novices wanting to start additional slow can lower this all the way down to 50mg and probably see
some useful results nonetheless. Advanced customers
typically increase the oral dose as a lot as 200mg every day,
keeping in mind that these doses are unlikely to current the risks of liver toxicity.
References:
Legalsteroids.Com Reviews
For Anavar, its major use is in chopping cycles where preserving
lean muscle mass is essential whereas lowering body
fats. It Is additionally favored for its minimal unwanted effects and decrease
threat of liver toxicity compared to other
steroids. Anavar (Oxandrolone) is understood for its mild anabolic
properties with low androgenic results. It is
often chosen for chopping cycles as a outcome of its capability to promote lean muscle mass
with out significant water retention.
Its solely respectable medical use originally was the treatment of very severe cases of weight loss because
of numerous diseases together with cancer and HIV/AIDS.
Anavar additionally has a reputation for being one of many
safest steroids to use, with relatively few unwanted effects reported.
What makes Anvarol the higher Anavar alternative
is its capacity to help you keep power and energy ranges whereas
on a calorie-restricted food plan. It does this by
rising ATP (adenosine triphosphate) manufacturing, which offers more vitality on your muscular tissues
to work with. As a result, Anadrol should solely be used by skilled customers who’re
conscious of the dangers involved. T helps enhance power ranges and strength, enabling
you to push through intense workouts.
This characteristic renders Anadrol less suitable for people aiming to attain a extra outlined, lean look.
Anadrol can cause unwanted side effects similar to high blood pressure, liver injury, and an increased risk
of creating heart problems. Subsequently, it could be very important seek the assistance of with
a healthcare skilled before beginning any sort
of Anadrol cycle. Anadrol is great for bodybuilders wanting fast muscle positive
aspects and energy. Users see big enhancements of their exercises, muscle progress, and even intercourse drive.
Getting the proper Anadrol dosage is key for bodybuilders who need huge features with few dangers.
Anadrol, scientifically named oxymetholone, is a strong anabolic steroid.
Beware of some complement suppliers advertising their merchandise under the Halotestin name
– these are not the AAS, and such suppliers could be misleading.
Look for the word Fluoxymesterone on the label to ensure you’re getting the genuine
anabolic steroid. Hair loss is not usually mentioned, doubtless as
a end result of Halotestin is used for such short intervals that hair
loss doesn’t become prominent. On the other hand, pimples could be extreme for some Halotestin users, and some discover it
popping up in strange places like the torso and the usual spots on the
face, again, and shoulders.
Anavar is a popular anabolic steroid that bodybuilders and athletes use
to advertise fats loss, lean muscle progress, and increase energy.
The active ingredient in Anavar is Oxandrolone, which is a synthetic version of the hormone
testosterone. Anavar is not typically recognized for its vital strength-enhancing
properties in comparability with different anabolic steroids like Anadrol or Dianabol.
Whereas it does have some anabolic effects and may contribute to modest strength features, its primary focus is on selling lean muscle mass and fats burning.
Both Anavar and testosterone can be efficient for slicing, but Anavar is commonly most popular
due to its gentle nature.
Due To This Fact, virtually all of the weight gained on tren shall
be lean muscle mass, making it a more aesthetically pleasing drug for some bodybuilders.
This leads to the event of lean muscle mass and increased energy.
In addition, Anadrol additionally inhibits glucocorticoid hormones, which are liable for muscle breakdown. This
makes Anadrol an efficient steroid for both bulking and slicing cycles.
One other comparability worth mentioning on this
part is between oxymetholone and Superdrol. Once marketed as a prohormone
in the mid-2000s, Superdrol is one other powerful bulking
steroid that may rapidly add mass and strength, making it very
close to Anadrol when it comes to performance.
These may be a variety of the most debilitating and physique-ruining unwanted side effects of steroid use and embrace bloating or water retention and gynecomastia.
You can expect nice dry positive aspects without estrogenic
unwanted effects except you embrace different aromatizing
steroids in your Anavar stack. Exceptional fats loss will be seen on this stack, and it’ll
come on quickly. Expect an increase in energy and endurance, but the side effects of Clen can hurt your exercise capability (lowering the dose is right if you’re sensitive to stimulants).
They are also not very hepatotoxic, which means they can be utilized for longer periods at a
time. HCG just isn’t really helpful for ladies as a PCT due to it probably enlarging
ovaries (26) and inflicting virilization (27).
Clomid can be not a positive PCT for girls, as it might trigger the ovaries
to turn out to be oversensitive.
However, whereas it’s thought-about safer than most other anabolic steroids, it’s not with out potential side effects.
Although rare, Anavar could cause virilization in women if misused or utilized in high doses.
Typical recommended dosage lies in the 5-10mg per
day range, and the usage period ideally should not exceed 6-8 weeks.
The addition of Winstrol in this stack will amplify general anabolism and lipolysis.
Gynectrol has been specifically formulated to
naturally soften away unwanted chest fats. In conclusion, each Anavar and Winstrol
have their distinctive advantages and downsides. It
ultimately depends on your individual objectives and preferences
when it comes to selecting between the two. The solely facet effect that might be attainable
with these supplements that you’re using is to cause pimples to take place
to a larger extent when figuring out. However even though this could be
a temporary facet impact that might be overcome with correct diet and exercise, it is nonetheless highly recommended that it’s averted.
Additionally, trenbolone isn’t estrogenic (failing to aromatize),
making it a dry compound and thus a very desirable slicing agent.
Bodybuilders typically opt for the injectable kind, with it being considerably cheaper and more potent than oral testosterone.
Dianabol is also liver poisonous, being a C-17 alpha-alkylated steroid, thus having
to cross through the liver so as to become lively.
References:
steroids Dianobol; tap.ngo,
Generally used substances for PCT include selective estrogen receptor modulators (SERMs) similar to Clomid or Nolvadex.
These substances help block estrogen receptors and stimulate the manufacturing of luteinizing hormone (LH), which
in flip stimulates testosterone manufacturing. By optimizing the synergistic effects of
Oxandrolone with proper training strategies and nutrition, athletes and health enthusiasts can unlock
their full potential and obtain outstanding results.
Anavar Dosage GuidelinesTo maximize the advantages and decrease potential risks, it
is essential to adhere to applicable dosage pointers when using Anavar.
The optimum dosage of Oxandrolone varies depending
on an individual’s experience, objectives, and tolerance degree.
Another good factor about oral testosterone vs. injectable testosterone is its speedy effects.
We see serum testosterone levels peaking in the bloodstream simply 5 hours after the
first dose of oral testosterone. This is opposite
to different oral steroids, that are C-17 alpha-alkylated
and have to be consumed on an empty abdomen for full effects.
This is as a end result of of most orals being fat-soluble
and thus susceptible to lowered absorption via the gastrointestinal tract.
Nonetheless, we have discovered that operating
greater dosages will cause extra pronounced unwanted effects, which
we are going to element in the unwanted effects part of this information (further down).
Sensitive individuals who’re susceptible to side effects with testosterone are likely
to expertise the same end result with Sustanon 250.
For those that have already cycled different testosterone esters,
there’s little difference between swapping those esters for Sustanon 250 when it comes to dosage guidelines and stacking choices.
With this in thoughts, shorter esters require regular injections (daily or each different
day), whereas longer esters, similar to enanthate, usually only require injections
a couple of times per week.
Zac transitioned from being lean to ripped, gaining noticeable amounts of muscle
whereas reducing his body fat share and becoming more vascular.
Trenbolone doesn’t aromatize and cause excessive estrogen ranges, unlike some other bulking steroids.
However, gynecomastia stays attainable because of trenbolone raising progesterone ranges.
This is due to it inflicting important cardiovascular strain due to a rise in cholesterol levels
(LDL).
As you might need noticed on this site – we are
sturdy advocates of natural bodybuilding and health. Additionally, if you’re a person with low
testosterone levels, Anavar can be a good possibility.
This is usually a specific downside for bodybuilders and
athletes who are already involved about their appearance.
In addition to being embarrassing, gynecomastia also can cause ache and
discomfort. This facet effect is most typical in teenage boys and younger men, as their bodies are still growing and are more
sensitive to the consequences of steroids.
Our patients usually implement post-cycle therapy following trenbolone use to revive the hypothalamic-pituitary-testicular (HPT) axis.
Some novices might make the most of an Anavar-only
cycle, with it posing less toxicity than different anabolic steroids.
However, Anavar nonetheless causes doubtlessly troublesome
side effects (which are detailed within the side effects section below).
Despite Anavar producing only average will increase in lean mass and
acute weight gain, it is distinctive for enhancing muscular power.
Consequently, powerlifters and fighters underneath our care have commonly cycled Anavar, enabling them to become stronger without having to go up a weight class.
By strategically incorporating additional
substances into your cycle, you can amplify the benefits of
where to purchase anavar and obtain even more vital gains.
Nonetheless, it is essential to approach this
practice with caution and consult with a healthcare professional
before experimenting with combination therapies. Energy Enhancement and Performance BoostThe utilization of Oxandrolone during a cycle can significantly enhance energy ranges.
Anavar works higher with dietary adjustments
and strict exercises that entail weight lifting and cardio.
Through its use, you’ll find a way to address unnecessary
deposition of fats for these, leaner, dry gains.
Taking into account the limiting capability to endure
steroids, women shouldn’t settle for in extra of 5 mg/day in the major week.
Individuals DO get sent to prison for taking steroids and if you’re doing dealings in the black market you’re running
the danger of going to jail and paying a hefty
fantastic. For those who don’t know, the main two attributes of anavar is to burn fat and enhance power.
Vital will increase in power can even assist in sparing muscle tissue when slicing (on low calories).
In future Anavar cycles, 20mg could also be used from the first
week onward; and the size of a cycle may be increased
to 8 weeks. It’s essential to notice that
virilization unwanted side effects are uncommon in ladies (who use moderate doses of Anavar).
And even if they did happen, they aren’t permanent in order soon as you cease taking
Anavar; your hormones will balance out and these male characteristics are prone to disappear.
This testosterone-suppressing impact nevertheless is prone to be mild, with
endogenous testosterone ranges usually recovering in a matter
of weeks or a few months.
Because of this interaction with aromatase, Trestolone displays therapeutic promise as a potential HRT
different, and will fill the function of a viable Check base
various in a cycle. Via these mechanisms, Trenbolone exhibits a a lot more sturdy inhibition of muscle protein breakdown than Testosterone.
In addition, it lowers corticosterone and cortisol
levels, while concurrently inhibiting cortisol from binding to skeletal
muscle glucocorticoid receptors [R, R, R]. General, Nandrolone
is far much less androgenic and estrogenic than Testosterone.
Nandrolone is also mildly estrogenic by itself via its ability
to behave as an Estrogen receptor alpha (ERα) agonist [R].
They are very anabolic, but additionally they exhibit significant amounts of satellite tv
for pc interplay with other receptors within the physique.
For example, Nandrolone is 5α-reduced into DHN, a a lot less androgenic metabolite, and it additionally interacts with Progesterone receptors within the physique which
can have an anti-androgenic impact on the body.
This means that it’s legal to own and use Oxandrolone with a valid prescription from a licensed healthcare supplier for accredited medical functions.
The duration of PCT can range from four to 6 weeks, relying on the the length of the
Oxandrolone cycle. ROIDTEST testing kits are quick, cheap, and accurate,
and embody the very newest advances in reagent testing.
Made with our unique STEROID ID™ technology for elevated accuracy and
reliability. The majority of the Anvarol evaluations from prospects are on the manufacturer’s official site, and they’re largely
very positive. Some of the evaluations ask questions about the product, while others rave concerning the potent benefits they received inside a couple of weeks.
To cut back the probabilities of gynecomastia occurring on testosterone, our patients often take a SERM such as Nolvadex.
Testosterone does not notably affect the liver, with it being injected
straight into the bloodstream. This can be attributed to Winstrol drying out the physique, resulting in less cushion for the
joints.
References:
does kali muscle do Steroids
Its effectiveness paves the finest way for considerable energy gains and lean muscle growth, setting it apart in the
saturated steroid market. Notably, the marginally higher price ticket
affirms the quality of this industry-leading product, with
a 50 capsule package, each containing 10mg, sometimes falling within the
$115-$120 range. Given the widespread influence
of testosterone within the body,, its decline can result
in noteworthy bodily and emotional challenges.
They don’t offer the bodybuilding and performance-enhancing results that
you could derive from utilizing genuine oral Anavar steroids.
If 10 milligrams per day, tolerated well with no virilization symptoms 50 milligrams
per day attempted. Anavar is a non aromatized steroid
which means estrogenic side-effects unimaginable. Nevertheless,anavar has a
DHT nature which can produce pimples, hair loss, and prostate enlargement.
Nevertheless, its mild nature will not yield as a lot
anabolic activity as its structural nature would suggest.
As of 2005, Anavar is available as a dietary supplement as
long as it doesn’t contain oxandrolone.
When discussing the potential professionals of purchasing Oxanabol, the listing is
really in depth. Are you considering to buy Anavar, the highly sought-after anabolic steroid known for its performance-enhancing effects?
Whether you’re an experienced athlete or a health fanatic trying to
take your coaching to the next stage, purchasing Anavar requires cautious consideration.
By following a structured cycle, customers aim to
maximize the benefits of Anavar while minimizing potential risks and unwanted effects.
Understanding the key parts and concerns of an Anavar cycle is crucial
for attaining desired outcomes and sustaining overall well being and well-being.
Moreover, Anavar stimulates the production of purple blood cells through a process referred
to as erythropoiesis.
Throughout these cycles, individuals ought to proceed to prioritize proper vitamin, intense
training, and sufficient relaxation for optimal
outcomes. It is essential to concentrate to potential unwanted effects and often monitor blood markers and total well-being.
It is essential to start out with a lower
dosage range, similar to 10mg per day, and steadily improve it to 20mg per
day if well-tolerated. Beginners should carefully
monitor their body’s response to the steroid and assess any potential side effects.
We are committed to providing you with the most dependable and unbiased product reviews attainable.
Our group of experts works tirelessly to analysis, check,
and consider products across varied categories, so you’ll
have the ability to confidently make informed buying
decisions. They take great satisfaction of their hands-on testing, thorough research process, and customer
suggestions analysis to ensure these reviews are reliable and informative.
We repeatedly replace the evaluations based on adjustments
in product availability or new data to guarantee
you have the most up-to-date suggestions. The pointers and recommendation in these
reviews are not substitutes for medical advice.
Please consult your physician earlier than taking any medicine or if you have doubts
about the product. The safest method is to
buy a legal anavar complement such as anvarol, which mimics
anavar’s effects and is FDA approved.
If you’re in search of a performance enhancement solution with out breaking the financial institution, Oxandro 10 from Magnum
Prescribed Drugs ought to prime your record. Oxanabol, delivered to
you by Alpha Pharma, has established itself as a frontrunner within the anabolic steroid market, paving
the way in which for athletes and bodybuilders to realize their fitness aspirations.
Its potency, effectiveness, and security profile make it an irresistible selection for users trying to witness
exceptional improvements in strength and lean muscle mass.
Anavar is a strong tool for anyone seeking to improve their physique, whether or not it’s for competitive bodybuilding, athletics,
or private health objectives. Anavar is a strong anabolic steroid identified
for its effectiveness in enhancing athletic efficiency and muscle growth.
Nonetheless, the effectiveness of Anavar largely depends on the quality of the product you’re utilizing.
Understanding its legal standing, sourcing it from trusted
suppliers, and adhering to recommended usage guidelines are crucial steps to make sure a responsible and efficient expertise.
Discovering dependable sources for Anavar in the USA is crucial to make sure product authenticity and
safeguard your health. With the proliferation of online marketplaces, consumers should method their buy
cautiously and observe particular tips to avoid counterfeit products.
There are seemingly numerous steroid suppliers that exist
on-line, and while many are price lower than the rubbish in your
garbage can, there are a lot of good ones. However, keep in mind any black
market purchase of Oxandrolone is unlawful within the
United States. You should get hold of a prescription to be able to legally purchase Anavar
and the purchase must be created from the pharmacy.
Many international locations around the globe carry
comparable legal guidelines, at the identical time many
are much more lenient; perceive the regulation because it pertains to where you reside earlier than a purchase
order is made. Regardless of where you live, if top quality anabolics are what you’re after and also
you wish to keep inside the safety of the legislation, you might be inspired to take a look at the sponsors right here at Steroid.com.
You will discover these sponsors carry top quality, high shelf
anabolics that may be purchased legally and not utilizing a prescription and
that present no concern as it pertains to a authorized violation.
Though Avanar and different courses of anabolic steroids
were initially designed to help individuals gain weight for medical reasons, today it has turn into the steroid of selection for bodybuilders.
Naturally, the hormone androgen is liable for male masculinity; nonetheless,
often some individuals wanting to look more masculine might resort to lab-produced androgen-like
steroids to boost seems. Bodybuilders depend on these
drugs to improve muscle buildup and weight.
References:
steroid stack for bulking
This aspect impact alone will be sufficient for so much of women to steer the selection to Anavar over Winstrol.
So although Winstrol is superb for fat loss, muscle retention, and bettering definition, its risk of unwanted
effects is much higher than Anavar, making
it a a lot less best option for females. Thus, we not often see customers acquire fat, water weight, or become bloated.
This is as a result of of Anavar being 5α-reduced; thus, it doesn’t aromatize,
which means estrogen ranges remain stable.
Do you finish up battling shedding fat, gaining a
lot of lean muscle, or having low testosterone levels?
Then Hi-Tech Prescribed Drugs Anavar is the complement resolution to buy for
you! These increased levels of free testosterone lead to extra muscle progress and so on. This process additionally inhibits glucocorticoid
hormones, meaning catabolic hormones, such as cortisol, are managed.
Buying Anavar online is normally a handy and cost-effective way to get the steroid.
The question I see most often requested by bodybuilders is how to Buy Anavar safely.
It’s always a good idea to seek the assistance of
with a healthcare professional earlier than beginning any new complement or medicine, together with Anavar.
They can present steering on whether Anavar is acceptable for you and might help monitor your health whereas taking it.
Sure, you can buy Anavar on-line from various
websites and online shops. However, it is essential to be
cautious and do your analysis to ensure you are shopping for from a reputable and trustworthy source.
PCT could be standard Clomid for 20 days – first 10 days at 100mg every
day, beginning two weeks from the end of the cycle. The lack
of threat of water retention is a significant benefit of Anavar and an enormous reason why it’s such a well-liked
and potent chopping compound, including for competitive customers.
You should not count on important muscle gains – Anavar
just isn’t a bulking steroid, but it might possibly promote some lean positive aspects
while concurrently losing fats. Women can achieve in the 10lbs range,
whereas males are likely to see smaller gains underneath
10lbs. Anavar excels at providing you with important improvements in aesthetics – leanness, hardness,
dryness, and, in short, getting ripped and shredded.
Anavar will help keep your performance and
strength capacity even on probably the most restricted of calorie-deficit diets.
You can maintain increasing once more relying on outcomes, restoration and the way you’re feeling.
The most dosage that could be safely consumed is 100mg everyday,
but that’s just for advance customers who are used to taking the drug and have
constructed up their tolerance. The identical could be stated for coming off
Anavar the place you slowly decrease the dose somewhat than just abruptly stopping and surprising your
body. It’s popular amongst bodybuilders as a outcome of anavar’s thought of a mild compound.
Although there are several side effects, because it’s not overly
powerful or poisonous, most of those unwanted effects can usually be prevented.
Like with different anabolic steroids, Anavar is unlawful in most nations, until you’ve a prescription from your physician.
Shopping For Anavar online presents convenience and a
broader selection of vendors. Whether Or Not you’re contemplating shopping for Anavar on-line or offline, this guide will equip you with the knowledge to make an knowledgeable and protected determination. I can’t mention sufficient instances that the higher the quality or purity
of your Anavar is, the less of it you’ll must take to get the desired
effects and results. However, most girls are more probably to discover that Anavar is the one that offers them the best stability of minimal unwanted side effects
and exceptional outcomes. Other countries are significantly less
strict relating to possessing Anavar in your
personal use. The UK, Canada, Denmark, Finland, Sweden, and Norway are just a few nations
with more relaxed anabolic steroid laws.
Whereas it has many benefits, you will need to use it responsibly and under
the steering of a licensed healthcare provider to keep away from potential unwanted side effects.
Home Provide is the leading on-line store within the US, providing superior-quality anabolic steroids at market-best costs.
Purchase Anavar on-line, together with other required medicine
on your bodybuilding and medical purposes, and avail quick supply.
Shopping For Anavar on-line is with out question the easiest approach to get this anabolic steroid.
There are seemingly countless steroid suppliers that exist online, and while many are value lower than the
rubbish in your garbage can, there are many good
ones. However, remember any black market buy of Oxandrolone is illegal
within the Usa.
Males on a cutting cycle with Anavar can count on an excellent fat
loss, and it will be fairly fast because Anavar is a quick-acting
steroid that you’ll solely use for eight weeks max.
Outstanding fat loss might be seen on this stack, and it will come
on rapidly. Anticipate a rise in power and endurance, but the side effects
of Clen can hurt your exercise capacity (lowering the dose is good if
you’re delicate to stimulants). Anavar will present the capacity to construct muscle and keep strength whereas weight-reduction plan. The hardness and dryness of Anavar gains and fat loss are among its strongest and
most fascinating benefits.
This makes Oxandrolone(Anavar)’s hepatotoxicity just about none, even at excessive doses, and has
the lowest hepatotoxicity of all oral anabolic-androgenic
steroids(AAS). When you purchase Anavar 50mg, you’re
getting top-of-the-line oral steroids for cutting, lean muscle preservation, and efficiency enhancement.
It’s a fantastic addition to any cycle, particularly for athletes and bodybuilders seeking to keep lean whereas
growing energy. It helps with fat loss, improves muscle definition, and helps muscle development
without causing significant water retention.
This was an excessive cycle duration, with a normal
cycle size of 6–8 weeks for men. From this research, we can conclude that natural testosterone
manufacturing is likely to stay fairly high if a
reasonable dose or cycle is performed. Anavar and all anabolic steroids are primarily
forms of exogenous testosterone; thus, Anavar will improve muscle mass.
Facet results are definitely potential, however
for the healthy grownup they can be minimized.
In truth, with responsible use, many will discover they expertise
no adverse results at all. In order to know the unwanted side
effects of Anavar, we’ve damaged them down into their separate classes with
all the related information you’ll want.
Anavar is greatly appreciated by many athletes for a
quantity of specific causes. This steroid will
enhance energy; the entire improve will not be as significant
as steroids like Dianabol and nowhere close to the extent of Halotestin, but will in all probability be notable.
Strength is among the major components of profitable athleticism as it translates into velocity and energy.
Whether Or Not you are seeking to bulk up or get ripped, Anavar is a steroid
that may assist you to attain your targets. Purchasing them from an unreliable
source can result in serious well being consequences,
so it is not worth the risk. It’s additionally necessary
that you do not take this medication in case you are pregnant.
This medication may cause start defects and probably hurt your unborn youngster.
You should make positive that you are using efficient contraception when taking this treatment
and let your physician know right away when you become pregnant whereas on this medication.
IJOHMR Journal is an Open-access, Peer Reviewed, Indexed, Bi-monthly journal publishing
analysis articles from varied sub-disciplines of Drugs and Dentistry.
References:
short do
Nonetheless, in apply, we find women expertise a quantity of signs of clinically low testosterone levels following
anabolic steroid use. Nonetheless, if a person stacks Anavar with different anabolic steroids,
this suppressing effect will be exacerbated.
Alternatively, if a person does not desire to wait several months, they will incorporate
post-cycle remedy to scale back this recovery time interval.
Anavar’s capacity to extend energy is principally
because of it being exogenous testosterone. Nevertheless, we discover it additionally has a dramatic effect on adenosine triphosphate manufacturing and creatine content contained in the muscle
cell. Higher levels of ATP are helpful for people wanting enhanced
power when bulking. It can be advantageous for users who’re cutting
and at risk of dropping power due to prolonged restriction of calories.
This can lead to a more practical workout
and dramatic muscle development in much less time. Alcohol will increase cortisol, a catabolic hormone
that may blunt a few of Anavar’s fat-burning and anabolic effects.
A liver help complement is crucial when stacking Anavar with different hepatotoxic orals, such as Winstrol, Anadrol, or
Dianabol.
This gave BTG seven years of market exclusivity, leading
to a pharmaceutical monopoly. Consequently, BTG was in a place to significantly increase the worth of Anavar by 1,200%.
Our team of medical experts are there for you in each
step of the method in which, from finding the best doctor and hospital to any type of assistance.
We educate and empower households to ensure that right healthcare selections are
made.
Known for its relatively gentle side effects and versatile performance, it
is usually utilized in both slicing and bulking cycles.
Users report impressive earlier than and after results, showcasing its
potential for reworking one’s physique with correct use
and adherence to pointers. Anavar, known scientifically as Oxandrolone,
is amongst the most versatile and popular anabolic steroids on the
market. Are you involved by the potential Winstrol results after 2 weeks
of use? Winstrol, also called Stanozolol, is a popular anabolic
steroid that’s renowned for its capacity to reinforce performance and promote
lean muscle positive aspects.
A dosage range of 5-10mg per day is typically considered applicable for female customers.
Trenbolone is a highly potent chopping agent that is identified for
its ability to increase muscle mass while selling fat loss.
When stacked with Anavar, it can help to amplify the muscle-building and fat-burning effects
of both compounds. This stack just isn’t beneficial for
novices, as Trenbolone is a highly poisonous injectable steroid.
Misuse or overuse can result in health points like liver and coronary heart issues.
It’s vital to have interaction in strength-enhancing substances like Winstrol responsibly, complying with suitable dosages
and cycle durations. The length of a Primobolan cycle may also rely upon the user’s
experience and supposed outcomes. It’s essential
to stability cycle length with relaxation durations
to take care of natural testosterone manufacturing and
overall health. Overall, cycling Clenbuterol may be an efficient way to improve fats loss, endurance,
and efficiency.
Both Anavar and Winstrol could be poisonous to the liver, however Anavar is generally thought-about to be
much less toxic. Anavar is a 17-alpha-alkylated steroid, which means it has been modified to cross through the liver without being broken down. This
can put extra stress on the liver, however Anavar
is much less poisonous than different 17-alpha-alkylated steroids.
Extra particularly, people with higher expertise in anabolic steroid use typically lean towards
the upper finish of this dosage spectrum. It is essential
to underscore the gender-specific considerations in Winstrol dosages.
Males and ladies typically reply differently to anabolic steroids,
necessitating a tailored method. This not solely considers the variance in physiological responses but additionally addresses potential concerns associated to virilization in girls.
Individualizing the dosage takes into consideration the varied
expectations customers might have, whether or not it’s to enhance muscle definition, enhance athletic performance, or obtain particular
physique objectives. The nature of the cycle, encompassing
elements like length and accompanying compounds,
also performs an important role in dosage willpower.
Winstrol is extra anabolic than androgenic, making it extra suitable for female usage; nonetheless, the dose should be maintained
low, between 10 and 20 milligrams at most.
Subsequently, oral Winstrol is superior for faster
outcomes, which could be skilled within the first week of
a cycle. Due To This Fact, injectable Winstrol is a greater
possibility for many bodybuilders, because it essentially amplifies results without having to increase the dosage.
Equally, we find injectable Winstrol poses less cardiovascular threat, as hepatic lipase isn’t stimulated, and HDL cholesterol levels thus don’t decline
significantly. Winstrol, on this regard, is the opposite of
Deca Durabolin, which can truly improve joint health as a outcome of elevated production of synovial fluid.
The advantage of water loss is a extra ripped look because of less extracellular fluid obscuring muscle definition. Furthermore, a lack of estrogenic conversion can exacerbate blood stress, with estrogen having a positive effect on ldl cholesterol.
Based on our lab outcomes, Winstrol is doubtless certainly one of the greatest steroids
for muscular endurance due to the stimulation of new purple blood cells (erythrocytosis).
Although steroids typically get negative press as a result of their misuse, there’s no denying the
essential role they play in bodybuilding. They mimic the hormone cortisol that our our bodies produce naturally in our adrenal
glands. The core perform of steroids is to lower inflammation and
suppress the immune system.
These testimonials illustrate the potential benefits of
the Anavar and Winstrol cycle, corresponding to improved
muscle definition, enhanced power, and increased athletic performance.
On the opposite hand, Winstrol or Stanozolol, operates similarly but with a
more pronounced impact. When Winstrol binds to the androgen receptors, it additionally stimulates protein synthesis and inhibits catabolism.
Nonetheless, Winstrol has a stronger affinity for the androgen receptor, leading to
more pronounced anabolic effects. One of the distinctive features of Winstrol is its capability to reinforce muscle definition and vascularity.
Anavar, known scientifically as Oxandrolone, works by binding to androgen receptors,
stimulating the cell to supply more protein.
References:
buy Anabolic steroid Online
If you’ve used steroids earlier than, you can start on the
greater end of the dosage range. We will also focus on the method to cycle Anavar, in addition to present recommendations on the way
to minimize the danger of unwanted side effects. Turinabol,
nevertheless, is a derivative of methandrostenolone
(Dianabol), which has very low androgenicity and isn’t a DHT-derived steroid.
Thus, we conclude that Turinabol is the safer steroid to soak up regard to preserving hair thickness and stopping recession. However, Anavar is a spinoff
of dihydrotestosterone (DHT), the hormone liable for hair follicle miniaturization on the scalp.
In one study, females were given as a lot as one hundred fifty mg per day for 30 weeks, and
none of them demonstrated any masculinization (3). Ladies may
even experience noticeable lean muscle features because of its positive impact on nitrogen retention and protein synthesis.
While Anavar offers quite a few benefits for fitness fanatics, it is crucial to concentrate on potential side effects and take
necessary precautions to ensure a secure and effective cycle.
Oxandrolone is known for its relatively mild nature, however
some people should still expertise unwanted effects.
These can include acne, oily skin, adjustments in libido,
and potential liver toxicity. It is important to notice that these side effects are generally dose-dependent and
can differ from individual to individual.
However if allowed to proceed creating through continued use of the
steroids, it could turn into so pronounced that the one remedy option might be surgical.
Steroids which have more powerful androgenic properties would be the steroids that may cause virilization quicker
and extra severely than steroids that have a
lower androgenic ranking. Some steroids are utterly off-limits to females because of this; they are too androgenically powerful to be of any constructive profit
to ladies. It’s critical to be aware that it’s not regular for blood to look when you’re injecting steroids into the
muscle. If there might be any blood whenever you insert the needle, you hit a vein or artery rather than simply muscle tissue.
The needle then have to be eliminated, and no solution ought to be injected; as an alternative, begin once more
and discover a new muscle spot that does not draw
any blood. It’s essential to get their suppressed testosterone again in normal working order.
Dragon Pharma is a leading authority in the realm of anabolic steroids and performance-enhancing supplements, dedicated to empowering
athletes, bodybuilders, and fitness lovers worldwide.
With years of experience and a commitment to high quality, Dragon Pharma provides scientifically-backed insights and cutting-edge product reviews that help people achieve their peak bodily potential.
Whereas there’s a lack of scientific research on the
topic, anecdotal research performed by bodybuilders reveals that
oxandrolone can actually be a muscle-building drug. Nonetheless, the quantity
of lean mass gained during an Anavar cycle will rely largely on one’s intercourse, diet, coaching, and if the drug is “stacked” with other steroids.
So judging Anavar results could be a murky proposition, but one which’s made simpler by understanding a lot of what has been written within the
bodybuilding community. That stated, take a glance at the next topics, a
lot of which draw from forum threads here at EliteFitness.com.
Regarding Anavar for girls, virilization, or the
development of male sex traits, can occur with greater doses of Anavar (19).
Higher glucose uptake helps sustained power ranges and prevents excess storage as
fat. This metabolic shift advantages athletes
needing optimum vitality management and people managing weight by way of
dietary interventions. The changes in cholesterol levels caused by Anavar can lead to an elevated threat of
heart illness, heart attacks, and strokes. Men who have already got cardiovascular
issues or are at risk for them should be significantly
cautious when contemplating Anavar use. Excretion rates depend upon renal operate, hydration standing, and urinary pH.
Impaired kidney perform can extend drug retention, increasing systemic exposure.
Anavar given to healthy men, has been shown to extend protein synthesis by as much as
44% and improve results of resistance training. Anavar can make you are feeling extra energized and powered up throughout physical actions.
This power increase, together with sooner muscle constructing, is what athletes are inclined
to search for, however it comes with unwanted effects
that could be extreme with long-term use. Anavar was designed for
short-term use, and medical sources, such as WebMD, level out that abusing
this drug with excessive doses and extended consumption can also cause
cardiovascular points [8]. Excessive blood pressure is likely certainly one of the
commonest medical situations on the earth, and it can result in quite so much of
serious well being problems.
One of the main reasons Anavar is so effective at burning fats is as a result of it helps to increase your metabolism.
Many patients don’t notice the toxicity of
prolonged alcohol abuse and how it impacts the physique.
Alcohol detox at the luxurious rehab habit centers at Gratitude Lodge leeches your physique of these toxins in preparation for
profitable therapy for medication and alcohol abuse. Alcohol detox might not
take as lengthy or produce extreme withdrawal signs, but it’s
nonetheless a vital beginning to your restoration.
It has been proven to provide spectacular results in users seeking to obtain a lean and toned physique.
It can be a valuable addition to the fitness
routine of both men and women who goal to improve their physical appearance
and efficiency. One of the most well-liked steroids in Canada, Anavar is also
one of the most secure anabolics obtainable. No androgenic side-effects makes this powerful steroid excellent for women as well.
Consistently delivering top of the range steroids and anabolics to Canadian athletes since 2012, Roidrx is your reliable and trusted partner in progress.
Anavar’s weak hepatotoxicity in regard to the liver is unique,
with it being an oral C-17 alpha-alkylated steroid.
So now we know it’s perfect for cutting that fats and getting a six-pack,
how do we use it for cutting? Simple, can be utilized alone at 60-80mg daily in males and 10-20mg every day in females for 5-7
weeks or together with different androgens that can complement its effects, More on that later.
When in comparison with other anabolic steroids, Anavar stands out for its ability to offer substantial advantages with a relatively mild aspect effect
profile.
You might use a bunch of compounds in this cycle, but one will usually
be the MAIN compound that takes on the primary anabolic role for
the duration of the cycle. The half-life of a steroid gives
you a way of figuring out how long that steroid will stay active in your system at a degree where performance and bodily advantages will be noticeable and achievable.
As Soon As you’ve injected, the ester begins to detach from
the hormone, and where it’s a long ester, it may possibly take every week or
two before the effects of the steroid kick in. It also takes longer to exit your system
when you cease injecting, and this impacts whenever you begin PCT.
By enhancing the effectivity of how your physique uses nutrients from meals (mostly carbs, proteins, and fats), every calorie you
take in is used to its full potential. Our information is designed to assist
give you the information you have to safely and effectively buy
steroids online within the United States.
Nevertheless, the approximate half-lives that are identified for all our commonly
used anabolic steroids provide a stable base to plan your
cycles on. Steroids play a role in optimizing the nutrient
pathways of the body by effectively shuttling vitamins to muscles which are giving the signal that
protein synthesis is being initiated21. So all these
high quality carbohydrates, healthy fat, protein, nutritional vitamins, and minerals are making their method
to the muscle tissue faster and extra considerably than if you have been not utilizing steroids.
Secondary steroids are used along with major anabolic medication to control unwanted effects or enhance the
effectivity of a cycle. These steroids play an integral part in maintaining hormonal balance,
regulating estrogenic exercise, and ensuring general well-being through the cycle’s duration. Therapy methods may embody
the use of medicine including aromatase inhibitors that block testosterone from converting to estrogen and SERMs, which assist decrease ranges of estrogen. Steroid use may be
essential to any steroid technique as it allows safer, more effective outcomes.
References:
steroid pills side effects
To enhance the effectiveness, combining the dosage with a protein-rich food plan and incorporating regular cardio and
strength training is crucial. This holistic strategy emphasizes the importance of
a tailor-made lifestyle to maximize the benefits
of the Anavar chopping cycle for women. It is important to
do not overlook that Oxandrolone is a extremely regarded and widely used medication known for its optimistic influence on sufferers affected by varied medical conditions.
Severe side effects may embody liver disease or toxicity, heart illness,
heart attack, or stroke.[1][2] Talk to your healthcare provider about
potential unwanted aspect effects and dangers.
These circumstances can happen with out warning or symptoms and might result in liver failure,
inside bleeding, cancer, or demise. Using anabolic steroid medicine may cause cholesterol (lipid) changes
within your blood, which can enhance fatty buildup inside your arteries (also called atherosclerosis).
Talk with your physician about the risks and advantages of using oxandrolone.
Oxandrolone, also known as Anavaros, is commonly utilized in athletics to sustain performance, protect muscle mass, and hasten recuperation between training classes.
An Anavaros regimen is greatest suited to people who have already got sufficient muscle mass and a moderate
degree of physique fats. Its major purpose is to boost muscle definition and scale back subcutaneous
fat, providing a more sculpted look to the muscle tissue.
There isn’t any denying that Anavar 50mg occurs to be one of
the most popular steroids you will hear people mentioning within the bodybuilding
industry.
For males, a typical dosage ranges from 20 to 50 mg per day, whereas girls typically use
decrease doses, around 5 to twenty mg per day, to minimize the risk of virilization. Because this is such a gentle and side-effect pleasant steroid it is often known as The Lady Steroid as a big portion of those
that purchase it are in-fact female athletes.
Anavar represents one of the well-liked oral anabolic steroids of all time, and this is largely because of its well-tolerated nature.
This is probably certainly one of the few anabolic steroids
that can be used safely by men and women, and it’s additionally
some of the side impact pleasant. Nevertheless, in some circles Anavar is
tremendously underappreciated because of its delicate nature, however this is usually due to unrealistic
expectations. Many tend to assume all anabolic steroids ought to
yield a set of particular effects at a particular rate of power, but
reality tells us varying steroids carry varying results
and functions.
Due to its delicate androgenic nature, Oxandrolone can be favored by feminine athletes who
search efficiency enhancement without the danger of significant virilization results.
Anavar (Oxandrolone) is a well-liked oral anabolic steroid that is recognized for its delicate androgenic properties and
highly effective anabolic effects. It is often utilized by bodybuilders and athletes to
help improve muscle mass and energy without causing excessive water retention or bloating.
Anavar is generally well-tolerated by most customers, with minimal unwanted
side effects when used responsibly.
Absolutely, the manufacturer Hi-Tech Prescribed
Drugs recommends users take Anavar for anyplace between 4-7 weeks with an 8 week off
cycle break between cycles. Hi-Tech also recommends taking a PCT or Post Cycle Support upon completing a cycle of Anavar.
Once a girl has built up some tolerance to Anavar, she could choose to begin future cycles at 10mg
per day and extend the cycle length to six
weeks (for further results). “Buy our weight reduction peptides and melanotan 2 peptides securely, with confidence, by way of our dependable on-line payment platforms.” “Efficient web site, order processing times and delivery with nice prices and products. Extremely advocate their web site and products.” “This was my first order to Roidrx and I’m fully stoked at the quality of the products and the promptness of the transport. I undoubtedly might be inserting my second order in a quantity of weeks.”
This prohormone is great for including in a dry prohormone to a cycle or used
alone to offset the environmental estrogens in your food regimen. Lastly, Anavar® makes use of Epiandrosterone, which is an endogenous steroid hormone found
in Anavar® with an androgenic exercise. Epiandrosterone and is produced in the body as a metabolite of
testosterone.
Long-term use or excessive dosages can lead to
more severe unwanted aspect effects corresponding to
liver harm, ldl cholesterol changes, and hormonal imbalances.
Common monitoring and session with a healthcare skilled are
suggested. Thus, bodybuilders who want to expertise
the advantages of anavar purchase it from the black market,
which is an illegal apply and should result in a fantastic or prison time.
If you’ve a history of heart assaults, hypertension, strokes or cardiac ailments, then you should neglect
about Anavar cycles.
This guide explores Anavar’s advantages, the way it works, and why Pharmaqo is a trusted supply for buying it in 10mg and 50mg.
Anabolic steroids are composed of testosterone merchandise (male intercourse
hormones) and they generally enhance muscle mass and improve
athletic performance. Even although anabolic steroids can be
misused and trigger unwanted side effects, they provide great scientific benefits when used therapeutically.
In this article, we current the best methods to search out steroids on the market.
Purchase Anavar 50mg (Oxandrolone) – the go-to anabolic steroid for athletes
and bodybuilders trying to reduce fats, build lean muscle, and enhance efficiency.
Identified for its gentle nature, Anavar is ideal
for sustaining muscle while shedding fat during a chopping cycle.
Whether Or Not you’re bulking or cutting, this steroid delivers real results with minimal side effects.
Unlike other steroids, Anavar preserves lean muscle while shedding excess fat, making it a staple for cutting cycles and performance enhancement.
The beneficial beginning dosage is 40-50mg per day, divided into two doses,
so that you simply can feel the impact of taking Anavar.
It isn’t suggested to take anavar buy in higher dosages or
for longer than beneficial. Oxandrolone in a solo cycle is ready
to provoke a reasonably sturdy enhance in weight but
only in newbies whose physique isn’t but used to using steroids.
Nevertheless, if you correctly combine Oxandrolone with different drugs,
you can mutually strengthen their effect and get an excellent outcome within the form of
an increase in body weight. Anavar is a nicely known steroid,
which is popular amongst bodybuilders and athletes.
The steroid is understood for its significant advantages, including enhancing
power, endurance, and muscle acquire.
In conclusion, when contemplating the decision to buy Anavar,
it could be very important approach it with caution, responsibility, and correct analysis.
Anavar can supply vital benefits corresponding to elevated muscle mass, improved energy, and enhanced athletic performance.
Nevertheless, it’s crucial to prioritize safety, authenticity,
and high quality when purchasing Anavar.
If you’re dieting and supplement with this steroid
you will achieve the leaner and harder look you need in a more pronounced method than you
would have with out. It is one of the few steroids that may lend to this finish equally in each men and women and as
it’s virtually always side-effect free it stays some
of the highly fascinating steroids of all time. Yes, you need a
prescription out of your doctor to buy Anavar within the United Kingdom.
This is as a result of Anavar is a strong steroid and can have antagonistic health
effects if taken without the supervision of a professional.
References:
anabolic steroids stacks
This is a steroid that will suppress your natural testosterone manufacturing quite significantly.
For this reason, you have to embrace a testosterone steroid in your cycle to keep away from falling into a low testosterone state.
Only a low dose of testosterone is required to
fill this function during your Masteron cycle, as you are not using that steroid for any
purpose other than as a testosterone alternative.
Thanks to its lack of aromatizing activity, Masteron doesn’t include a number of
the unwanted effects that steroid users most frequently dread of
an estrogenic nature.
These unwanted effects may be highly uncomfortable, impacting daily activities and quality of life.
Customers need to be ready for the bodily and mental pressure that
Tren can cause, as its potency makes it difficult to handle.
The severity of those unwanted effects usually requires cautious monitoring and, in some instances,
medical intervention, highlighting the importance
of warning when using Tren. For people which might
be cutting and weight-reduction plan down, Trenavar is ideal because ordinarily when you’re losing fat, you additionally expertise
a loss of strength too.
Trenbolone excels in simultaneous muscle achieve and fats loss
because of its versatility. Other anabolic steroids could possibly match trenbolone’s anabolic nature or
fat-burning attributes alone, however not simultaneously.
As we already talked about earlier, one Trenbolone acetate facet impact (much like many other steroids) is its power to scale back your body’s pure testosterone manufacturing and suppress your testosterone levels.
Since Trenbolone is incredibly well known and is one of the most powerful anabolic steroids on the market, it’s no surprise that the thought
of tren bodybuilding is very tempting. Trenbolone’s highly effective results
can result in psychological dependence, where customers feel
they can not obtain their goals with out it. This dependency might delay use, heightening the danger of unwanted effects and health problems.
It’s delicate when used alone, and honestly, you won’t
find many bodybuilders relying solely on Deca. It’s essential to notice that the severity and prevalence of those side effects can range from individual to individual.
Moreover, the use of trenbolone without medical supervision and in excessive doses can increase
the chance and depth of those unwanted effects.
The Drug Enforcement Company (DEA) explains that anabolic steroids are Schedule III substances beneath the
Controlled Substances Act.
Most noticeable is its lack of exercise
on the aromatase enzyme, which means we get no estrogenic effects, which leads Masteron to
be such a good hardening and drying agent.
These properties additionally enable Masteron to behave equally to
an aromatase inhibitor, although not as powerful as the actual thing.
Still, it might go a way in helping stop gynecomastia when stacked with aromatizing steroids.
These are merely the Drostanolone compound with a unique
ester connected to manage the release fee and the steroid’s half-life once in the physique.
As a outcome, the physique burns extra calories, which finally ends up in a discount in physique fats.
In terms of fat-burning, Trenbolone has been proven to improve
physique composition and enhance fat burning. Trenbolone can cause liver toxicity, mood swings, and psychological well being points similar to anxiousness and
depression. A typical PCT for women includes human chorionic gonadotropin (HCG) and a selective estrogen receptor modulator
(SERM) similar to Clomid or Nolvadex. Nevertheless, it is essential
to use caution when stacking and to keep away from combining steroids which
have related unwanted side effects.
Masteron will provide a pleasant psychological increase
to this cycle, drying you out and including vascularity.
Muscle features may also be reasonable; count on 10 lbs on the most, however
this ought to be totally maintainable. Many men declare their disappointment that
Masteron is such an unbelievable steroid in each different method apart from its capability
to remove your hair literally. “Confident, assertive, happy”
is what Masteron can do to our psychological outlook.
This may be because Trenbolone might help athletes practice longer,
get well quicker, and build lean muscle mass rapidly. For those
looking to naturally assist testosterone levels post-cycle,
dietary supplements such as Redcon1 Growth Stick Testosterone Help present safer options to sustain muscle-building progress.
Moreover, some anabolic steroids can have androgenic results, additional complicating sexual operate.
Nevertheless, its use in humans is illegal without a prescription due to its potential for abuse and dangerous unwanted effects.
While its use is predominantly male, some women are exploring trenbolone to succeed in their fitness aspirations.
However, earlier than diving into this controversial substance,
it’s essential to weigh both the potential advantages and
dangers involved. Research suggests that our
bodies turn into resistant to the results of testosterone
over time. In quick, taking Trenbolone will boost your testosterone ranges momentarily,
but then injury your body’s natural production post-cycle.
Many discovered that when taking testosterone
that their increase in muscle mass would get even smaller for each additional improve in dosage.
Dependancy risks, legal consequences, and strain on relationships add to
the perils of steroid misuse. Anabolic steroids may cause many problems
corresponding to cardiovascular which includes hypertension, left ventricular hypertrophy, impaired diastolic filling, polycythemia and thrombosis (7).
The effects of steroids on lipid profile embrace reducing in Excessive Density Lipoprotein (HDL),
growing in Low Density Lipoprotein (LDL) and complete ldl cholesterol.
These modifications are leaded to increase the risk of atherosclerosis within the coronary arteries (11-13).
Indirectly, increasing the concentrations of LDL as a outcome of abuse of anabolic steroids may lead
to an increase in sensitivity of platelets (14).
References:
where can i get steroids to Build Muscle
As a health coach, I am devoted to helping people achieve their well being
and wellness targets. With a robust background in exercise science and
years of experience, I design personalised workout plans and provide steerage on nutrition to maximise
results. Ultimately, the choice of how a lot Trenbolone to
use weekly ought to be dictated by your distinctive circumstances.
Deliberate thoughtfulness and consistent monitoring of your body’s response to the substance will ensure that you find your optimum dosage.
Heed your internal compass; it will information you in hanging the optimal balance
between health progress, muscular development, and
overall well-being. Figuring Out the appropriate
dosage is a vital a part of incorporating it into
your fitness regimen. Several essential factors come into play when deciding what’s best for
you.
We have discovered that one method to overcome insomnia is to eat ample quantities of steak, rooster, or turkey earlier than bedtime, combined with large quantities of carbohydrates.
These three types of meat are rich in l-tryptophan, an amino acid that has sedative qualities, serving
to to assuage users’ central nervous methods. Combining l-tryptophan with carbohydrates in a meal aids absorption and amplifies its soothing results.
This just isn’t considered critical and is more than likely associated to trenbolone’s excessive androgenicity, causing
inflammatory lipids (prostaglandins) to turn into activated.
Thus, upon injection, vasoconstriction of the muscular wall (in the bronchus) causes some irritation to the lungs.
We have found that respiratory distress can even occur when injecting different steroids, corresponding to testosterone cypionate.
Pumps shall be extreme throughout exercises, however this will also be a common theme outside of the health club
due to increased glycogen storage.
After utilizing Winstrol, the kind and size of PCT males will principally depend upon what other AAS you’ve stacked it with.
Winstrol is most sometimes utilized close to the top
of a cycle, and few cycles will use it as the primary compound.
This means most suppressive effects will doubtless come from different AAS being used.
We use a lower Winstrol dose right here due to Clenbuterol’s
potential adverse effects on the guts.
Thirdly, trenbolone can have some side effects, but these are usually dose-dependent and can be easily managed with correct supplementation and coaching.
Secondly, trenbolone just isn’t a “magic bullet” and it won’t magically rework you right
into a bodybuilder in a single day. Trenbolone can cause side effects corresponding to hair
loss, pimples, gynecomastia, and liver injury. Trenbolone is an artificial steroid that
was first created in the Nineteen Sixties. It is a
19-nor derivative of testosterone, and it has each anabolic and androgenic properties.
However, it’s usually considered harsh and not advisable for first time users.
It’s primarily beneficial for intermediate and experienced customers as a result of its robust nature.
Sometimes customers could be having an underlying health condition that may
worsen with using Sustanon. It is always essential to get a check-up before you begin utilizing any steroid,
just to make certain. By using an excellent PCT (post
cycle therapy) complement, you’ll be able to help to scale back the chance of many of these unwanted effects.
Most bodybuilders, health lovers, and athletes who use Sustanon 250 cycle report experiencing lowered recovery time between exercises.
When you cycle these substances, you’ll have the power to improve
your total energy whereas additionally reducing the
chance of side effects.
Many of those people are first-time customers of Clenbuterol, and it is known that those who are using
Clen for the primary time are more likely to suffer from unwanted effects.
One of the reasons Clenbuterol is not approved for use in the USA is
its potential to cause everlasting organ damage.
Research have discovered that a really high share of Clenbuterol customers who
experienced extreme unwanted effects ended up having to be admitted
to hospital. Extended use at greater doses can take Clenbuterol’s safety to all new ranges,
and this is the place the risks will amplify significantly.
Trenbolone and Deca Durabolin are both injectable bulking steroids,
but they are completely distinctive in their pharmacology and results.
We have discovered Equipoise to be more estrogenic than Deca Durabolin, with EQ displaying 50%
less estrogenic effects of testosterone but 400% greater than Deca Durabolin. Studies indicate that Deca Durabolin is
mostly well-tolerated by ladies when taken in dosages of a hundred mg
(6), administered each other week for 12 weeks (thus translating as
50 mg/week). Although Deca Durabolin will not be essentially the most optimum steroid for
women (with Anavar being a extra suitable
compound), analysis suggests Deca Durabolin presents a low danger of virilization. We find this cycle better suited to someone nervous about gynecomastia, as an aromatase inhibitor (AI) can be taken, synergistically decreasing
estrogen and progesterone ranges. Thus, by stacking Dianabol with
Deca Durabolin, customers is not going to be significantly aggravating these two symptoms, with Deca being an injectable and having much less toxic effects
on blood lipids.
Combining these essential efficiency enhancements takes exercises
to new ranges, with many users able to push via previous personal greatest lifting limits.
These performance boosts can be substantially improved by stacking Testosterone Enanthate with a few of the
more potent steroids like Dianabol. Testosterone Enanthate is probably
essentially the most extensively used steroid as it’s
broadly obtainable, inexpensive, and very effective for performance enhancement.
Most bodybuilders will inject Testosterone Propionate every other day, making for a three-times weekly administration. This injection can cause lots of muscle
soreness and sometimes irritation, so you have to rotate the
injection site every time. Health authorities often warn in regards to the
potential for long-term health problems as a
result of regular, high-dose anabolic steroid use.
Dr. Jack Parker, holding a Ph.D. and pushed by a deep ardour for health, is a trusted skilled in bodily well being
and authorized steroids. He blends thorough research with hands-on expertise to assist Muzcle readers obtain their health
targets safely and effectively. Outdoors of labor, Jack loves spending time
along with his household and maintaining with the newest
well being tendencies and analysis. Before using Trenbolone,
it’s essential to consult a healthcare skilled. A doctor or
healthcare provider can help you understand
the potential dangers and unwanted effects of
Trenbolone use, as properly as provide guidance on dosages and cycles.
Additionally, they can monitor your health and
intervene if any negative unwanted effects come up.
In either case, trenbolone and Winstrol can be extraordinarily
efficient when used together. If you wish to bulk up with out putting your well being in danger, then a
safe and legal Trenbolone alternative is your best bet.
These alternate options are designed to imitate the consequences of Trenbolone without any of the harmful side effects.
In Addition To the obvious well being risks,
there is additionally the authorized threat of utilizing Trenbolone.
It’s made with all-natural components and has been shown to be just
as effective at serving to customers bulk up. When used correctly, trenbolone may help you achieve the physique you
desire much faster than when you have been to coach alone.
There are lots of myths and misconceptions about what
trenbolone does to your body.
Since it’s sturdy, introducing this compound to the system results
in a response. Apart From these two, another type is known as Trenbolone Hexahydrobenzylcarbonate (Tren Hex).
It has a half-life of ten to fourteen days and is a long-acting steroid.
Moreover, it’s necessary to keep in mind that no two bodies are similar.
What works completely for one particular person could not work the identical means for an additional.
References:
weight loss steroids for men – output.jsbin.com –
This enhanced androgenicity can successfully forestall erectile dysfunction.
Deca increases progesterone, an identical feminine hormone to estrogen, and
thus puffy nipples may be experienced by some users. This is a
comparatively low dose and most of my pals who cycle lots have been telling me how my take a look at was too low, take
a look at must be minimum 500, and so on. I by no means saw any profit
from going excessive test, for my part test is only there to keep you from turning into lethargic and likewise to keep your sex drive up…
At 175mg/week test and 350mg/week tren my sex drive was fucking amazing,
gf needed to tap out a quantity of times, and my gainz? Lol at this low dose I was KILLING my good friend
who was on like 500mg deca, 800 take a look at. As we’ve already
mentioned, abusing Tren can include loopy side effects.
Former residents of one complex mentioned Tren de Aragua grew emboldened last summer season, when gang members realized management had effectively
abandoned the buildings. In Aurora, legal exercise
among Venezuelans centered round three apartment complexes.
The buildings, all owned by New York-based CBZ Management,
had been uncared for for years earlier than giant numbers of
Venezuelans moved in, metropolis officers mentioned.
If you hook up with WiFi on trains, in stations, and at resorts,
it’s wise to get a VPN to stop snooping in your knowledge.
I use and advocate NordVPN which is fairly priced and tends to work nice; it additionally
lets you simulate visiting web sites from pretty much wherever on the earth.
There are plenty of tunnels in Spain and WiFi cuts out completely each
time a quick prepare goes by way of a tunnel. Ave, Avlo, Ouigo, and Iryo
are all comparatively dependable and most of the time, you presumably can assume that your high-speed practice will depart and arrive on time.
Whereas some international locations have accredited
veterinary applications, Trenbolone enanthate cannot be obtained legally and
is simply available on the black market. Obtaining it from an underground
lab can additionally be off-limits and no form of
Trenbolone has acquired approval for human clinical use.
Therefore, it goes without saying that Trenbolone should not be thought of
an option for feminine bodybuilders or any other type of
user.
Trenbolone or tren is commonly used recreationally by bodybuilders, in the slicing phase of bodybuilding prep.
This is as a outcome of highly effective anabolic results, mixed with the
anti-catabolic effects and prevention of visceral fats accumulation. Nevertheless, because of the lack of
proof and litany of potential unwanted side effects, the risks far
outweighs the profit in trenbolone use. Security, efficacy,
and therapeutic dosage, cannot then be decided
to the absence of well established and proven evidence.
The recommended dosage for Tren varies relying on the
person’s experience with anabolic steroids and their particular goals.
It Is essential to begin with a low dose and steadily improve it to keep away from potential unwanted
effects. If you resolve to use AAS like trenbolone acetate, achieve this cautiously and with as low a dose
as possible.
Possession or use of Tren with no prescription can result
in authorized consequences, impacting users’ personal and
skilled lives. Additionally, using Tren in aggressive sports activities may violate
ethics or rules, damaging reputations and leading to disqualification. The authorized and moral considerations surrounding Trenbolone make it a high-risk possibility for individuals who prioritize their public picture and adherence
to regulations.
Whereas experienced steroid users shall be much less delicate via years of cycles and can experience milder gains.
We generally see androgenic side effects from this stack, as
tren and take a look at each have sturdy androgenic properties.
This could also be in the type of oily pores and skin, pimples, hair loss, or difficulty urinating.
Athletes and bodybuilders tread a fine line when figuring out the optimum dosage to attain their desired results while safeguarding
their well being. Two major kinds of trenbolone are commonly used,
trenbolone acetate (also often known as tren ace) and trenbolone
enanthate. This quick, higher-dose Tren cycle is suitable for blasting earlier than a comp.
An intermediate Trenbolone consumer would never feel the want to
stack Tren with some other compounds except for Check.
Sadly, trenbolone also can trigger low testosterone levels as properly
as numerous unwanted effects including nausea, high blood
pressure, hair loss, and pimples. Quite A Few anabolic steroids have unfavourable traits, such as high aromatisation ranges.
Trenbolone, in contrast, does not achieve this, which is considered
one of its benefits. Oestrogen encourages the growth of fat cells, which outcomes in larger male breasts and extra physique fat.
Oestrogen additionally slows metabolism, which makes
it much harder to shed weight and much easier to realize it.
Trenbolone’s dose is raised, while Masteron compliments Trenbolone to offer the supposed lean, shredded, and tough look.
Masteron is utilized by competitive bodybuilders only
in the course of the pre-competition periods. So, it’s an official veterinary
drug used principally for reinforcing cattle
mass to acquire meals.
While the positive aspects won’t be as large, they are going
to be high-quality dry gains which are simpler to hold up.
There isn’t any scarcity of adverse feedback about Tren cycles,
though typically, it will consist of men just talking about a number of the worst unwanted
facet effects and tips on how to handle them. Some more unfavorable critiques naturally heart on Tren’s tendency to have an effect on your psychological functioning.
A higher dose during a chopping cycle of 200mg-300mg per week can introduce
more problematic side effects, notably on the psychological side
and issues like night time sweats and insomnia. In my experience, it’s
ALWAYS ideal to begin using Tren at a lower dose the first time, even when you’ve got plenty of
experience with other AAS and don’t contemplate yourself
a newbie.
Nonetheless, if you’re already somebody who suffers from a short fuse and common irritability,
Tren will make it considerably more pronounced.
You’ll then have to assume about this impact on your work
and personal relationships to determine whether or not Trenbolone is worth utilizing for you.
The preliminary trigger is regarded as when the oil-based
injectable solution inadvertently reaches tiny capillaries, permitting access to the bloodstream.
Trenbolone is acknowledged for its ability to speed up muscle progress as a end result of its potent anabolic properties.
It enhances nitrogen retention within the muscular tissues, which is
essential for protein synthesis and muscle restore. This process allows muscles to grow sooner and stronger than they’d naturally.
Many bodybuilders report seeing results within just a few weeks, making
it extremely engaging for these seeking to bulk up shortly.
Unlike different steroids, Tren’s muscle-building results are seen in a quick
time frame, creating significant positive aspects which are often difficult to attain without dietary supplements.
Regardless of where your Trenbolone Enanthate doses
fall, so as to maximize use the compound should be injected two instances a
week. Simply take your total desired weekly dose and
cut up it into two smaller equal sized injections, similar to one on Monday and the
opposite on Thursday.
References:
rich piana steroid (Tyler)
These are merely the Drostanolone compound with a special ester hooked up to control
the release price and the steroid’s half-life as quickly as
in the body. The ester itself does not affect Masteron’s activity because it turns into hooked up over time once you’ve administered the steroid,
leaving Masteron to take full impact. Enanthate is more difficult to find,
even though it comes with the profit of fewer common injections due to its
longer half-life. Masteron is an injectable steroid and
some of the extremely regarded AAS used for chopping and getting shredded.
The aesthetic results you possibly can achieve with Masteron are dramatic
and spectacular, making this a favorite steroid within the bodybuilding
and physique competitors.
Trenbolone raises levels of bradykinin, an inflammatory mediator
peptide that helps blood vessels dilate. The bradykinin peptide is widely known for
inflicting a cough response in these taking ACE inhibitors for hypertension. Seasonal fluctuations within the value of Tren Ace may happen due to factors such as changes in demand, availability of
raw materials, or promotional offers from manufacturers or retailers.
It’s necessary to regulate market developments to establish potential
value variations. By contemplating these
elements beyond worth, you may make a extra complete assessment of the worth for cash offered by Trenbolone Acetate.
Keep In Mind that while price is an important consideration, it shouldn’t be the only real determinant of worth.
The mechanism of motion of Trenbolone involves its
interplay with androgen receptors in the body.
Trenbolone is an artificial spinoff of the hormone testosterone and exhibits sturdy anabolic and androgenic
properties. When administered, it binds to androgen receptors located in numerous tissues,
including muscle cells. Trenbolone is a strong anabolic steroid that has been linked to hair
loss and untimely baldness in users. The actual mechanism by which Trenbolone causes these problems just isn’t totally
understood, however it’s considered as a end result
of its masculinizing effects.
Primobolan is understood for its capacity to advertise lean muscle
mass and improve general performance without inflicting significant unwanted aspect effects.
Nonetheless, I additionally discovered it to be fairly expensive, making it much
less accessible for those on a decent price range. When it involves utilizing Trenbolone
Acetate, it is important to comply with a well-planned cycle to maximise your positive aspects and minimize any potential unwanted facet effects.
A typical Trenbolone Acetate cycle lasts between eight and 12 weeks and is usually combined with different steroids
for even greater positive aspects. When it comes to dosing and administering Trenbolone Acetate,
it’s important to begin slowly and progressively enhance the dosage over time.
This will help to minimize any potential side effects and
permit your body to regulate to the steroid.
Some will run Masteron for just four weeks, but I discovered that’s simply if you
begin seeing its most results, so I prefer to go slightly longer to 6 or 8 weeks.
200mg per week can present all the effects you want for a
pre-contest cycle, usually used in the last weeks of an extended cutting or lean bulk cycle.
Newbies or these operating Masteron alongside TRT can benefit from as little as 100mg/week.
Once more appropriate breast most cancers medication had been developed, the use of Masteron declined and ceased by the late Nineteen Eighties.
If unwanted facet effects become severe or persistent,
it’s essential to consult with a healthcare skilled. Bear In Mind, the most secure method
is to keep away from anabolic steroid use altogether,
as even with precautions, risks cannot be utterly eradicated.
When embarking on a trenbolone cycle, it is
crucial to prioritize security to reduce potential well being
risks and maximize outcomes. This section outlines essential security measures
that customers ought to implement during their trenbolone cycle.
Trenbolone is, subsequently, illegal for any use as an anabolic steroid and is topic to sanctions by
all world sports activities anti-doping organizations.
Incredible energy and pumps are a standard
function of any optimistic Tren critiques. This improves
workouts, and you will feel like every workout is improving
in a single space or another. This means elevated weights, more
reps, and greater capability to work out properly past
your regular level. Trenbolone is amongst the most talked about steroids, and a lot of users won’t draw back from sharing their own experiences and results with this steroid.
In contrast, probably the most familiar ester of Trenbolone
Acetate is a veterinary product for use in livestock
within the US to help within the development of extra muscle mass in the months before they’re slaughtered.
As A Outcome Of of the widespread use of Trenbolone
Acetate within the beef cattle trade, it’s a
steroid that is not tough to return by. On the downside, comparable unwanted side effects to different
steroids are nonetheless possible as a end result of Tren is a progestin, which, sorry to say, also can make you more vulnerable to gyno.
Additionally, trenbolone use can lead to increased aggression, which could be problematic for some individuals.
It is essential to notice that these unwanted facet
effects aren’t skilled by all users, and some might expertise
them to a larger degree than others. It is important
to comply with a correct post-cycle therapy (PCT) to
help your physique get well after a cycle of Trenbolone.
The truth is that Tren places a slight pressure on the liver
due to its resistance to most of the liver features such as breaking down and excreting
hormones similar to insulin and bilirubin. The rise
within the degree of estrogen leads to the enlargement of a man’s
breast. In the lengthy term, you won’t be able to achieve your objectives
as a result of your bodily and mental situation is not good.
The stress of staying awake for an extended time will eventually affect your physical and mental health.
When the chemical construction that tells the body to sleep or to wake up is destroyed, you expertise insomnia.
Nonetheless, they might leave scars on your face that can be troublesome to get rid.
Pharmaqo is a quantity one steroid brand, offering high quality products to fitness
center fanatics for years. Pharmaqo’s support team can assist with questions, ensuring a easy
purchasing expertise and high quality assurance. As one person has mentioned, “a little little bit of voice cracking near the end of the cycle” signifies an onset of virilizing signs.
It’s clever for any female to cease taking Primo
at that point (unless virilizing effects aren’t a concern).
But as I’ll detail later, the choice between which form to
take comes right down to extra than simply your choice for swallowing a tablet or injecting.
It can additionally be essential to notice that this steroid
can inhibit glucocorticoid hormones, which is essential for stopping muscle
catabolism. Trenbolone Suspension is a 19-nortestosterone (Nandrolone) by-product that’s suspended in a water-based
answer for injection. Tren suspension is nandrolone with 2 added double bonds inside the nucleus.
It is a steroid that has high anabolic and androgenic results of 5
occasions greater than testosterone. It has no estrogenic
activity but does possess high progestogenic activity.
For physique related functions, most males will discover a dose of mg each different day to be a fantastic vary.
50mg each other day is a unbelievable place to start out
with 100mg each other day typically being all the
Trenbolone Acetate many men will ever want.
A standard stack consists of the usage of a testosterone ester and Winstrol for the previous few weeks of a cycle.
An 8-16 week cycle is appropriate for a Tren enanthate cycle at a hundred to 300mg weekly for a newbie to intermediate consumer.
Skilled users of Trenabol enhance the dosage to 500mg per week, either
in a quantity of injections or, in the case of extremely skilled individuals, in one
single injection. Trenbolone has an enormous status among bodybuilders as the last word anabolic steroid, but its historical past is relatively unremarkable in comparability with many other AAS.
References:
best illegal steroids
The different major objective when formulating Anavar was to create a gentle drug with few
unwanted effects so women and kids could safely take it.
It is not very toxic and has been used to deal
with youngsters that suffer from extreme burns so as
to construct body mass which has been lost, to quicken restoration. By
utilizing Anavar cycle the consumer can reduce fats and due to this fact physique weight whereas retaining muscle mass.
If you wish to go for summer season holidays on the beach you can take Anavar for slicing fatty tissue
in the physique while gaining energy. The choice to purchase Anavar on-line in UK brings your fitness
aspirations inside attain. With the potential to boost muscle growth, enhance endurance, and obtain a more outlined physique, Anavar
on the market caters to a diverse array of fitness aims.
The comfort of purchasing Anavar on-line
empowers you to embark on your fitness journey confidently, understanding that you simply’re accessing a robust
device favored by athletes and fitness fanatics worldwide.
Anecdotal evidence suggests that people love how Anavar
helps them feel strong and look good without the extra bulk.
The delicate enhancements in muscle tone and composition permit users to benefit
from the mirror as a lot as the weight room. The market provides a selection of
Anavar options, with brands like Maxtreme Pharma, Dragon Pharma, and Alpha Pharma leading
the sector.
An effective PCT protocol can accelerate the recovery of
endogenous testosterone. Moreover, water retention is unlikely on Anavar, with the compound making a dry and defined look.
This diuretic impact can be more doubtless to improve vascularity and muscle striations while contributing to a leaner look.
Our expertise and scientific studies indicate that elevated liver enzymes generally return to normal following cycle
cessation (11). Based on our tests, Anavar is probably certainly one of the best steroids in regard to toxic unwanted effects.
Analysis has additionally shown it to possess
safety in long-term medical settings (9). Anavar has a significant influence on energy, despite being a slicing steroid and not causing dramatic weight gain.
There are notable variations in how Anavar impacts male and female customers, as well as variations
in beneficial dosages and cycle durations. Anavar’s mild nature makes it female-friendly, and girls usually discover PCT
unnecessary due to its mild impression on hormonal
steadiness, particularly with reasonable doses and shorter cycles.
The body’s natural equilibrium can typically restore
itself post-cycle, considering the common monthly cycles in ladies.
Advanced customers may think about 60-80mg per day, however
this choice requires cautious monitoring of their body’s reactions.
Greater levels of ATP are useful for individuals wanting enhanced
strength when bulking. It is also advantageous for
users who are chopping and susceptible to dropping power due
to extended restriction of energy. We have had sufferers report vital strength results on Anavar, even when consuming low calories.
This can be why powerlifters usually administer Anavar previous to a contest for optimum
strength with out significant weight achieve.
In the world of bodybuilding, bodybuilders use Var for his or her bulking, chopping, and efficiency goals.
Anavar can be utilized alone or with other oral and injectable androgenic-anabolic steroids (AAS).
Anavar is fascinating for fats loss, so your
food plan will be a high priority when using this steroid, and it will make or break your outcomes irrespective of how efficient Anavar is as a steroid.
As I talked about, Anavar is among the few steroids girls can confidently use.
Cycles should be restricted to six weeks, and the dosage must
be 10mg daily.
In comparison, Anavar comes with a far higher anabolic
rating but a a lot decrease androgenic score of simply
24 – which implies its androgenic exercise is significantly milder than testosterone.
Females can acquire energy and lean positive aspects
at low doses, and stacking Anavar with another kind of compound referred to as Ostarine (a SARM) can bring about excellent outcomes with minimal
side effects. It comes with some advantages over Anavar, however ultimately, your
targets would be the decision maker on which to choose.
Masteron is an injectable steroid, and it’s the safer one to make
use of for longer cycles. Anavar at 10mg/day for 6-8 weeks is nice, and you most likely won’t have any unwanted effects.
As for the most effective time to take Oxandrolone, cut up it
into two doses day by day at 5mg/each (once with breakfast and again before bedtime).
It’s essentially the most tolerable and the least more likely to cause virilizing signs at low to moderate doses.
Nitric oxide (NO) sports vitamin has turn into the most important sports vitamin product class.
But, despite dozens of “developments” in NO supplementation, traditional NO sports diet still remains flawed for one elementary reason. This flaw can simply
be detected by simply inspecting the formulations in NO products across-the-board
all through the sports vitamin industry. Arginase is the enzyme that
breaks down arginine, which is an issue for NO supplements because arginine is the source of NO manufacturing.
With the exception of Hi-Tech Prescription Drugs, no different complement company has discovered tips on how to sort out the arginase downside head-on…
Most male rivals could have between 3% and 5% body fat
throughout competitions. Remember that these are probably the most excessive
customers, and they are probably to make use of
different compounds alongside or as a substitute of Anavar.
As you can see, estimating a simple lack of fat in pounds is virtually inconceivable.
Nonetheless, due to the presence of trenbolone, we consider this to be a harsh cycle and
not suitable for novices. Anavar’s capacity to increase strength is
especially as a end result of it being exogenous testosterone.
However, we discover it additionally has a dramatic effect
on adenosine triphosphate production and creatine content contained
in the muscle cell.
References:
where can i buy anabolic steroids
When it involves combining Anavar with different substances,
caution is suggested. Anavar is understood to be a comparatively delicate steroid with low potential for
side effects, however it could possibly still be dangerous when used
irresponsibly or in combination with certain substances.
Yes, we ship Anavar to Australia, making certain bodybuilders
and fitness lovers get genuine products with quick supply.
Oxandrolone for sale is supplied within the form of tablets which might be taken orally.
Take it at the similar time every day in order that you do not forget to take
oxandrolone on-line. If you’ve been bedridden (incapable of walking)
for a protracted time while using this medication, seek the assistance of
your doctor. It is also used to reduce muscle loss caused by steroid use and to scale back
bone ache in individuals with osteoporosis.
Constructive reviewers will state that Anavar
is essentially the most well-tolerated steroid they’ve used.
Still, it’s necessary to take a glance at what dosage they’ve used so you
can determine how the dose would possibly influence your results and
(lack of) side effects. This permits us to gain perception into
how different people expertise Anavar. This offers you a noticeable increase in power, in addition to increased
endurance and stamina.
By increasing nitrogen retention, Anavar supports an environment favorable
to muscle growth and repair, particularly useful throughout intense coaching durations where recovery
is important. Even though this drug is quite well-liked with girls, there is evidence that those who take Anavar can expertise sudden adjustments of
their menstrual cycles [12]. (4) Magnolini, R., Falcato, L., Cremonesi,
A., Schori, D., & Bruggmann, P. (2022, July 17).
It is favored by each women and men as a end result of its gentle
yet effective nature. Nevertheless, correct dosage, cycle size, and PCT
are essential for protected and effective use.
Like other anabolic steroids, Oxandrolone carries potential risks if misused.
However, it’s considered one of many milder and safer steroids when used responsibly
underneath medical supervision. Common unwanted side effects may embody liver toxicity, acne,
testosterone suppression, and virilization symptoms in ladies.
The fats loss mixed with lean gains can outcome in an general achieve in physique weight, however importantly,
it shouldn’t be water weight, and the features ought to be
all muscle. Enhanced muscular definition is possible after a single Anavar cycle
for women, even at lower doses of 10mg. Anavar is among the many
top-rated and most beneficial steroids available in the marketplace
for athletes and bodybuilders. The reason it is liked by many,
is as a result of it’s capable of triggering the expansion of the muscle
mass with as few unwanted effects as possible, making it one of many safest to use.
I’m not saying Lazar 100 percent took anavar, however if you diet onerous
and work your butt off, these outcomes are typical of somebody taking anavar for 8 weeks.
Not to say that anavar will maintain your muscles trying
fuller on-cycle. You can go onto Youtube and watch customers focus on their first expertise and listen to folks debate about what they suppose
and the way a lot they’ve taken and for how
lengthy. There’s a bunch of stuff you could
read about and see how others have responded to taking
Anavar and what product they’ve used. Although there are barely varying opinions it appears that the recommend amount of time
to take Anavar for is around 2-4 months (8-16 weeks).
Anyone with a historical past of liver issues must avoid Anavar completely.
It is banned by the FDA and several sporting federations due to it being
a growth-promoting compound and its antagonistic results.
Although some girls could expertise fewer virilization effects with Anavar compared to different steroids, particular
person responses differ, and there can nonetheless be significant risks concerned.
There are a handful of high quality manufacturers on the market, but many
are under-dosed or mislabeled. Some low grade manufactures will actually label the
steroid Oxandrolone but they’ve truly used cheaper Dianabol powder.
The particular person still receives a strong anabolic impact, but the complete effect just isn’t what he was after.
Subsequently, one strategy would be to have an AI prepared
if you begin to notice swollen nipples. As soon as our patients start
to experience the early levels of gynecomastia, they begin operating letrozole on the dosages
said above. This is undesirable information, considering blood stress
is already prone to be excessive during this cycle.
This metabolic shift benefits athletes needing optimal vitality management and individuals managing weight through dietary interventions.
Anavar can make you’re feeling more energized and powered up during bodily activities.
This power boost, along with sooner muscle building, is
what athletes are probably to look for, nevertheless it comes with unwanted effects that can be
extreme with long-term use. Anavar was as quickly as used
to promote development in youngsters, however now development hormones are more
generally used for this function. Oxandrolone is used to help you regain weight lost after surgical procedure, extreme trauma,
or chronic infections. Oxandrolone is also used in individuals who
can not gain or keep a wholesome weight for unknown medical causes.
Not Often, males might have a painful or extended erection lasting
four or extra hours. If this occurs, stop using this drug and get medical
help right away, or everlasting issues might happen.
Circulating testosterone as the hormonal basis of sex differences in athletic performance.
Furthermore, if an individual has experienced gynecomastia, this is a sign that the substance is Dianabol (42).
These are some of the points bodybuilders who visit our clinic face when buying UGL Anavar.
Equally, utilizing Anavar all 12 months round on a decrease dose compared to
administering the next dose within the brief time
period, the person is less likely to notice outcomes.
Concurrently, it safeguards muscle tissue from catabolism, enabling people to realize a more defined and ripped physique.
Powerlifting is a demanding sport that requires strength, power, and endurance.
Anavar, with its capacity to advertise muscle growth, increase energy, and cut back body fat, could be an efficient supplement for powerlifters.
SARMs are generally thought of safer than traditional steroids, as they aim particular
tissues and sometimes have fewer side effects. Safer options like the best oral steroid with
least side effects or safest steroids for over 50 ought to at all
times be prioritized. We thought-about security scores based mostly
on effects on ldl cholesterol, hair loss, and general unwanted effects.
This reduces the chance of estrogen-related unwanted effects, making them safer.
Anavar is commonly cited as the most secure oral steroid
due to its mild profile. Masteron won’t have an incredible amount of side effects, however some people have reported hair loss
to be outstanding while on Masteron. HGH does have unwanted aspect effects, corresponding to decreased insulin sensitivity,
water retention, and maybe carpal tunnel syndrome.
References:
most powerful legal Steroids (https://www.livebinders.com)
Whereas Anavar could assist in your bodybuilding journey, the street to a sculpted physique
nonetheless primarily depends on dedicated coaching, strong diet, and sufficient rest.
Don’t rush the method, and ensure any adjustments to your dosage
are thoughtfully thought of. When it comes to the usage of
Oxandrolone, commonly often identified as Anavar, understanding the suitable dosage is important, and
it can differ based on gender. Each men and women seek the benefits of Anavar for enhancing their
physique and performance, but the dosage requirements can range significantly.
In the desk below, we present a comparison of recommended Anavar dosages for women and men. Some
girls might expertise noticeable unwanted facet effects even at low doses, whereas others could tolerate
greater doses with minimal points.
For women, a daily dose of 10 mg will be incredibly helpful for lowering physique fats and building muscle mass.
Anavar is likely one of the best anabolic steroids to make use of
if you’re attempting to set new personal records for strength in the health club,
because it causes its users to experience incredible levels of power.
Typically, what most athletes and bodybuilders are hoping to perform by using
steroids is to enhance their muscle tissue during the bulking cycle.
They try to tone their muscle fiber, enhance energy and do away
with the excess fat. This cycle is for these already at a relatively low
body fats degree (average weight or below).
15lbs of weight loss is possible, depending in your meals consumption and train routine.
An interesting conflicting impact with these two compounds is that
Clen can lower your urge for food whereas T3 can enhance it.
This is especially true when it comes to oral steroids, as they
hydrate the cells with a higher affinity than injectable Steroids.
It is important to remember that simply since you aren’t bloated such as you could be on Dianabol
does not mean the compound isn’t working. It is a a lot leaner
compound than others, and you will only acquire dry and lean muscle.
DHT steroids can not work together with the aromatase enzyme to convert into Estrogen.
Therefore, taking Anavar can enhance the risk
of arteriosclerosis and will virtually certainly elevate blood pressure in all users.
If high doses are taken, cardiovascular unwanted effects can turn into severe, resulting in hypertension, coronary heart disease, or stroke.
Ldl Cholesterol ranges are likely to return to normal as soon as users discontinue supplementation. Other benefits of Anavar embody enhancing stamina and power because it
boosts pink blood cells. The joint benefits of Anavar make it
stand out above other cutting steroids and is one of my favorite things about
Anavar. Regardless Of its popularity as a extremely effective steroid,
it’s nonetheless a relatively gentle oral in contrast with other compounds.
Inexperienced users will share their unfavorable Anavar
experiences, but so usually, this revolves around the reality that a person didn’t use a testosterone base
when on an Anavar cycle! If Anavar is the primary steroid you
want to use, you’re still nearly actually going to
stack it with no less than a type of testosterone as a testosterone substitute during your cycle.
One of the less spoken-about advantages of Anavar is a attainable boost to your overall mood.
The similar can’t be mentioned for a lot of other steroids, which
may potentially trigger anxiousness and increased anger
instead.
Another notable good thing about Anavar is its capacity to assist with fat loss and enhance physique composition. This is achieved by way of its capacity to reinforce lipolysis, the process of breaking down fat molecules within the physique.
Users might discover a decrease in body fat and an increase in lean muscle
mass when following an Anavar cycle. In combination with a well-balanced food regimen and exercise routine,
Anavar may help people obtain a more outlined and toned
physique.
Nevertheless, they should be checked frequently on Winstrol (due to stronger hepatotoxicity).
Fuller muscles are also advantageous in competitions the place dimension is
rewarded. Muscle pumps on Anavar could be prominent, particularly in the
decrease back, with a few of our patients describing this sensation as painful when performing
deadlifts. This impact creates an outlined and full look on Anavar, whereas on Winstrol,
a toned appearance is widespread, however with much less of a pump to the muscle tissue.
Muscle fullness is possibly the only area the place
Anavar beats Winstrol relating to the advantages of each steroid.
The chemical buildings of Winstrol and Anavar are related, with each compounds being modified forms
of dihydrotestosterone.
Nonetheless, if you are new to steroids, it is always a good suggestion to begin with a lower dosage and see how your body reacts.
Some users report feeling more aggressive or irritable
while taking the steroid, whereas others experience a lift in confidence and temper.
However, these effects usually are not universal, and
a few folks might not notice any mood modifications in any
respect. In summary, Anavar can affect your libido by rising or decreasing your testosterone ranges.
One in style stack contains Testosterone Enanthate, also referred
to as Test E, and Anavar. One of the primary advantages
of Anavar for girls is its capability to promote lean muscle features with
out causing vital water retention or bloating.
This means that the features made during an Anavar cycle are usually lean and sustainable, somewhat than momentary
will increase in water weight. When it involves pure weight reduction or fats loss PEDs, Clenbuterol is considered
possibly the very best of the bunch.
Many ladies in the health trade have benefited from this weight reduction help
for its fat-blasting, muscle development, and strength-boosting capacity and comparatively milder impact on reasonable usage.
Anavar additionally ensures a fair risk-to-benefit ratio which isn’t normally the case with other bodybuilding steroids.
Once More we are speaking about average consumption or short-term
use of this steroid here.
This might not sound like a problem, nonetheless, women can report considerably much less well-being and a lower libido post-cycle.
Anavar is primarily used as a chopping steroid, so if a woman’s objective is to construct large amounts of muscle, different steroids would be extra beneficial (such as Anadrol).
Muscle features will be reasonable, serving to to
subtly enhance muscularity, with out trying bulky. Dr. O’Connor
has over 20 years of experience treating women and men with
a historical past of anabolic steroid, SARM, and PED use.
He has been a board-certified MD since 2005 and supplies steerage
on harm reduction methodologies.
Sure, Anavar can have an result on your period by
inflicting irregularities as a end result of its impact on hormone ranges.
People who want to acquire Anavar ought to consider buying from respected sources for authentication and quality.
The greatest place to purchase Anavar is thru the CrazyBulk
official website, as that would make sure the legality
and security of this merchandise.
References:
steroids Are A Type of
This step helps ensure that your usage aligns with private health needs and avoids
unnecessary risks. Inform your healthcare provider
about all drugs, together with prescription and over-the-counter drugs, and
dietary supplements to avoid potential interactions with Anavar.
When used under the supervision of a healthcare
skilled and as prescribed, Anavar may be secure.
Inexperienced users will share their unfavorable Anavar experiences,
but so often, this revolves around the fact that
a man didn’t use a testosterone base when on an Anavar cycle!
Few steroids may have us closely taking a look
at both male and female cycles, but Anavar is an exception. At All Times keep this in mind when deciding on your own Oxandrolone dosage.
Whereas Anavar is poisonous to the liver5, as we would
expect with an oral steroid, its hepatotoxicity degree is minimal in contrast with many
other steroids, making this a perfect alternative for novices.
Men and girls have separate doses when taking anavar, as they have completely different quantities of pure testosterone.
Men typically will typically take 15-20mg of anavar per day, with girls taking 5-10mg.
This permits bodybuilders and average gym-goers the chance
to reinforce their physique without taking excessively dangerous substances.
Anavar also possesses a second alteration at the 17th carbon place
by the addition of a methyl group that permits the hormone to be ingested orally officially classifying Anavar as a C17-aa anabolic steroid.
In conclusion, you will want to understand the risks and advantages of buying Anavar for sale in Mexico.
While it might be a more handy possibility for some, it comes
with the potential for counterfeit merchandise and authorized
implications. Yes, it’s attainable to purchase Anavar on-line in Mexico from various on-line retailers.
The hardness and dryness of Anavar positive aspects and fat loss are among its
strongest and most desirable benefits. Not having to deal with water retention is a aid for anybody wanting to achieve a shredded, exhausting, and vascular physique.
Endeavor a strict calorie-deficit food plan puts you at danger of muscle catabolism and losing power.
They could possibly prescribe Anavar in the event that they really feel
it’s applicable in your needs. Nonetheless, it’s
necessary to notice that not all doctors are comfortable prescribing steroids, so you may want to do some
research to search out one who is. First off, there are 3 main functions that Hi-Tech Prescribed Drugs
Anavar is well versed in that nearly all different merchandise have a tough time doing.
These functions are the potential to spice up
nitrogen retention, decelerate SHBG and obstruct glucocorticoid hormones.
I am a muscular man with a lot attention and recognition for my physique.
I started with very little and always put 110%
into the fitness center to get to the place I am now.
From anabolic steroids to SARMs to peptides and ancillary drugs,
I’ve done it in some unspecified time within the future in my life, and I can relate.
Whereas there are upper and decrease limits of safe consumption of the steroid, the dosage will
range amongst people. Give your fitness targets a boost with UK
Steroids, essentially the most trusted place to
find Anavar for sale in the UK. Enhanced restoration permits an athlete to extend coaching
intensity.
Testosterone ought to be included in a complicated Anavar (Oxandrolone) cycle, even when at just a maintenance dosage of 100mg weekly with your chosen ester, with propionate or
enanthate being widespread selections. A whole vary of different steroids are
generally stacked with Anavar, including the highly effective fats burner Winstrol, Equipoise, Proviron, Masteron, or
Primobolan. This is not a priority for ladies who can proceed with an Anavar-only cycle on the beneficial 10mg day by day dose.
Most cycles will utilize testosterone past the 8-week
Anavar cycle size, extending to 12 weeks with testosterone earlier
than beginning post-cycle therapy. Anavar excels at providing you with significant
enhancements in aesthetics – leanness, hardness, dryness, and, in brief,
getting ripped and shredded. Anavar will help maintain your efficiency and
energy capacity even on probably the most limited of calorie-deficit diets.
Firstly due to the amount of Anavar on the market for human use
is so rare. Secondly, it’s often extraordinarily expensive in comparability
with different Anavar merchandise and lastly because of counterfeits
and faux merchandise supplied. Counterfeits exist as a end result of high price of raw Oxandrolone powder though manufacturing is affordable (tablets).
Though Anavar is offered as a pill and is less prone to spoil, you
could need to increase the effects of Oxandrolone by conducting a mixed cycle with one other injectable drug.
If a few ampoules arrive damaged, you will not have the flexibility to
get a refund of your money. Feminine Oxandrolone customers will usually dose the steroid at 5-10mg
per day. It’s not frequent for a woman to need anymore than 10mg per day,
but if more is required it might be increased by 5mg.
In fact, with accountable use, many will find they experience no unfavorable results in any
respect. In order to grasp the unwanted effects of Anavar, we have damaged them down into their separate classes with all of the associated information you’ll want.
It is always really helpful to purchase from a good supply and to consult with a healthcare professional earlier than beginning any
steroid cycle. Additionally, you will want to observe correct dosing
and cycle suggestions to attenuate the chance of antagonistic
results. Our suggestion is to stay within the confines of the regulation and try anvarol
(legal anavar). Anvarol is produced by a well-known company called Loopy Bulk,
who have obtained 1000’s of optimistic reviews.
If you wish to buy Anavar for personal use, you
are able to do so from some abroad suppliers, but you should take caution as these is probably not
legitimate sources. In this doc, we’ll provide an outline of Anavar’s legal status within the UK
and talk about how to purchase Anavar safely from abroad suppliers.
For those unaware, each week we now have a specific steroid or
PED up for dialogue.
Right Here at Best Value Vitamin you will find the most price effective value on this highly
effective supplement. The larger the dosage goes, though, the extra likelihood
of women experiencing antagonistic effects from Anavar’s androgenic exercise.
These androgenic results are generally recognized as virilization, or the attainment of masculine
features, and may embody a noticeable deepening of the voice and development
of physique and facial hair. Others find that testosterone suppression is a lot larger than anticipated.
Unexpected water retention is one other occasional adverse
comment customers make about Anavar – however these people are
nearly certainly victims of buying counterfeit Anavar.
As you can see, estimating a simple lack of fats in pounds is nearly inconceivable.
References:
oral testosterone steroids for sale [Quyen]
Nevertheless, Anavar is the exception to this rule, displaying powerful strength-building attributes; despite not
causing vital weight achieve. Bulking steroids are normally probably the most highly effective steroids for increasing
energy, with slicing steroids being inferior.
Anavar is not classed as a bulking steroid for that
reason, as its muscle-building results are inferior
to other traditional bulking steroids. Therefore, if somebody needs
to gain some muscle, but not an enormous amount of mass, Anavar
will do just that. Some women and men who do not wish to
take steroids, because of their dangerous results, may
decide to take an Anavar-only cycle, solely because of its safety profile.
There are a handful of high quality brands on the
market, but many are under-dosed or mislabeled. Some low grade manufactures will actually label the steroid Oxandrolone however they’ve actually used cheaper
Dianabol powder.
With Winstrol being a C17-alpha-alkylated steroid, it’s going to cause liver pressure while
reducing HDL cholesterol and raising LDL by way of the stimulation of hepatic lipase.
All of this, combined with a lack of aromatization, will cause potential injury to the guts.
We have discovered common cardio workouts to be helpful in stopping large spikes in blood pressure.
When it comes to using Anavar, males ought to approach the drug with warning as a result of its potential unwanted side effects.
However, if used responsibly and in accordance with
recommended dosages, Anavar can be an efficient tool for attaining specific fitness goals.
Overall, Anavar dosage for athletes must be rigorously considered and monitored to attain optimal results
whereas minimizing the danger of potential unwanted side effects.
In Search Of professional guidance and using Anavar responsibly can help athletes enhance their performance and obtain their goals.
You can stack Anvarol with different CrazyBulk
supplements that can help you attain your fitness objectives even faster.
Famend for its light influence, Anavar shines brightly, providing
customers a journey marked by its effectiveness and safety in achieving desired
outcomes. Embark on a journey the place the magnificence of Winstrol converges with the renowned efficacy of
Anavar, giving rise to the enchanting synergy often known as ‘Winvar’.
This fascinating combination emerges as a cornerstone for
these sculpting their physique in the course of the cutting section.
This profile makes Anavar one of many few anabolic steroids safely tolerated by feminine
athletes when used responsibly (Basaria, Journal of Gerontology).
Regardless Of having totally different paths, each Winstrol
and Anavar lead to elevated bodily efficiency. With Winstrol, users might anticipate important fats loss, improved power usage, and enhanced muscle definition.
In comparability, Anavar leans more towards sustainable energy positive aspects, elevated energy,
and enhanced performance without substantial weight features.
Thus, that is primarily a cutting cycle that can produce potent fat-burning and
average lean muscle features. The scales won’t improve as
they will on testosterone, however the lipolytic (fat-burning)
results might be extra outstanding.
By exploring the advantages, correct usage, and accompanying strategies, we aim to equip you with useful insights to embark on a
successful Oxanabol journey. Discover the potential
of this exceptional steroid and witness the transformative power it holds for girls looking
for spectacular physique enhancements. Oxandrolone a singular oral anabolic steroid hormone that’s also recognized as Anavar.
Anavar was first synthesized in 1962 and has been used within the medical therapy of catabolic disorders for over 30 years.
This characteristic implies that trenbolone
doesn’t convert to estrogen, so customers don’t experience water retention or
fats accumulation during a cycle. Trenbolone is actually an injectable steroid used by bodybuilders to
achieve large amounts of lean muscle and power whereas enhancing fat loss (1).
For the feminine performance enhancing athlete, whatever the
function of use, 5-10mg per day is generally the proper dose.
If extra is desired and 10mg per day has been well-tolerated, 15mg per day
can be attempted the following go round. However, every improve in dosing will improve the
danger of virilization. Doses of 20mg per day will strongly improve the danger with doses
above this mark all however guaranteeing some stage of virilization. Of the attainable unwanted effects of
Anavar essentially the most regarding will encompass cholesterol.
Anavar, a preferred anabolic steroid, has gained a reputation for providing
a strong answer tailor-made specifically for the female physique.
It offers quite a few benefits, such as lean muscle growth, power enhancement,
and fat reduction, making it a beautiful choice for those keen on sculpting their dream body.
A well-structured Check Anavar cycle can supply numerous advantages for individuals seeking to
optimize their gains whereas minimizing side effects.
By understanding the suitable dosages and durations, each women and men can tailor their cycles to swimsuit their particular targets.
Newbies ought to method steroid usage with caution, gradually growing dosages and closely monitoring their progress.
We will also cowl the latest analysis and news associated to Winstrol, so keep tuned for informative and engaging content material.
Bold decisions typically result in extraordinary results, especially in the bodybuilding circuit.
Deciding on a new stack can be difficult, with the many options out
there and the plethora of opinions on which is greatest.
An intriguing stack known for its potent results and dramatic outcomes –
it’s a pairing that’s brought on fairly a stir within the bodybuilding group.
Anabolic steroids are very effective for building muscle mass; however, it is very important understand the function that genetics, nutrition, and exercises additionally
contribute to a person’s transformation. In 1989, researchers discovered that 54% of competitive male bodybuilders in Kansas
and Missouri were taking steroids regularly, with 10% of the feminine bodybuilders
additionally admitting to such.
Nevertheless, Anadrol is a really poisonous oral steroid, straining the liver and the heart.
Subsequently, although outcomes may be useful by way of muscle measurement and strength, unwanted effects will
also be intense. Deca is one other bulking steroid that can enhance muscle and energy
gains. This is partly because of Dianabol being a potent oral steroid,
which is notorious for worsening cholesterol levels as it stimulates
hepatic lipase in the liver. Dianabol, being an oral steroid, will cause liver toxicity; thus, it should not be taken for an extended period.
Our patients often take a liver support complement (such as TUDCA) to
forestall ALT and AST enzymes from rising
too high. Thus, we discover androgenic side effects to be notably less on Dianabol; however, it is barely extra efficacious for muscle and power
features.
A heart-healthy food regimen, regular cardiovascular train, and consistent monitoring may help mitigate these effects (Glazer, Medical Science in Sports and Exercise).
Supports pure testosterone manufacturing, balances adrenal output, and helps reestablish hormonal
rhythm. Correct PCT after Anavar ensures that you simply maintain your hard-earned features, normalize hormone levels, and defend long-term health.
Food Plan was tremendous clear throughout this time and simply did about 2,300-2,500 energy a day with
2 grams of protein per physique weight.
References:
how to get steroids online
Some girls may need to go additional and add another two weeks to the cycle while
rising the dosage for those final two weeks up to 20mg day by day, however monitor for any potential unwanted effects.
Lean muscle gains will vary considerably amongst
girls, however over a 6-week cycle, some females can see wonderful features of up to 5-10lbs,
even whereas shedding some body fat. Testosterone Enanthate is a long-lasting ester that can be used for a 12-week cycle alongside Anavar.
This isn’t a priority for ladies who can proceed
with an Anavar-only cycle at the beneficial 10mg day by day dose.
Most cycles will utilize testosterone previous the 8-week Anavar cycle length, extending to
12 weeks with testosterone earlier than beginning post-cycle remedy.
Diet and present body weight will decide how far your fat loss can go, but a 5lbs
loss of fat over a cycle when you’re already lean will enhance the physique.
However, some other compounds are also utilized by females, together with Winstrol and Primobolan, in addition to the fat-burning
compound Clenbuterol. Anavar isn’t probably the most suppressive steroid, but
your natural testosterone manufacturing is prone to have taken successful.
This can cause symptoms of low testosterone when your cycle ends, together with loss of muscle and fats achieve.
If you have not any current kidney problems, utilizing low doses and short cycles
of Anavar is unlikely to cause kidney damage. Still, if you use
excessive doses and lengthy cycles, you’ll be putting your kidneys
– and other organs – at great danger of harm. Using
Anavar at low to reasonable doses is about as secure as it could possibly get for anabolic steroid use.
Purchasing Anavar for sale in 25mg or 10mg by way of PrecisionAnabolics is handy,
discreet, and delivered quick to your door, so you can start reaping the benefits in your
subsequent coaching session. These are solely some of the great advantages that might be gained from the right use of Oxandrolone (Anavar).
– It has been discovered to be protected for human use with no
extreme unwanted facet effects. The Impact of Location on Anavar Costs The cost of Anavar significantly differs from one region to
a different, shaped by native provide and demand dynamics.
These types of product promoters exploit the popularity of Anavar to market their own items, which lack
even a tenth of the effectiveness of the true Anavar on the market elsewhere.
Like different anabolic steroids, Anavar is not suggested for people underneath the age
of 21. Read about how celebrities use steroids and their favorites by clicking the hyperlink beneath.
You might be surprised to learn that anabolic steroids are incessantly
employed in conventional medicine to effectively treat a myriad of ailments.
For this reason, we’ve made a complete information to assist individuals like you buy Anavar on-line with out falling sufferer to all of the untrustworthy individuals
flooding the web.
However, this weight gain is primarily because of muscle progress and never water retention, making it a preferred alternative for a lot of athletes.
When it involves enhancing power without a vital enhance in mass,
many athletes flip to Anavar. This highly effective compound works
by promoting the synthesis of creatine phosphate in muscular tissues,
permitting for larger strength gains without
causing water retention. This is especially useful for athletes who need to remain within a selected weight class while
bettering their performance. When it comes to
constructing muscle and reaching a lean physique, Oxandrolone is
one of the hottest performance-enhancing supplements available on the market.
Whether Or Not you’re trying to shed weight or
acquire power, Anavar can provide a lift in your health journey.
For PCT, use 50mg/day of Clomid for three weeks, and you must recover rapidly.
All The Time keep this in thoughts when deciding on your own Oxandrolone dosage.
Past the standard issues, partaking in illegal transactions to acquire Anavar can have severe authorized consequences.
In this blog submit, we are going to delve into the necessities of buying
Anavar online, including the best manufacturers to consider and essentially the most reliable supply
to buy from — MisterOlympia.shop.
Liquid types of Anavar might provide sooner absorption due to their liquid
state, potentially leading to quicker onset of results.
Some users choose this type for its versatility, as it might be easily mixed with other liquid supplements or beverages.
However, it’s essential to train warning when utilizing liquid Anavar to ensure accurate dosing, as measuring
liquid volumes could be much less precise than merely taking a capsule or tablet.
As a end result, Anavar has less unwanted effects than the majority of steroids.
You will not expertise the same unfavorable effects as with different steroid types since this capsule does not convert to estrogen or DHT.
If you determine to use this medication, don’t count on it to have the identical
effects on you because it does on males. This is
because almost all steroids are stronger
in men than in ladies. With the rising fame of Anvarol, a quantity of shops at
the moment are retailing the product. While you should purchase the product from
other stores, to make sure that you’re shopping for the real product and also qualify for different
benefits, we recommend shopping for from the manufacturer’s web site.
The supplement is manufactured by CrazyBulk, a laboratory centered on researching
and producing steroid alternate options.
Whereas Anavar is a Schedule III controlled substance under the Managed Substances Act,
its legal availability in the US requires a prescription for medical circumstances like muscle wasting or extreme burns.
Nonetheless, it’s broadly purchased on-line, with many merchandise
labeled as Anavar typically containing substitutes like Methenolone or Oxandrolone blends.
This highlights the significance of sourcing Anavar from respected suppliers with product verification techniques in place.
The army generally doesn’t test for anabolic steroids, as it is sometimes an costly check.
As An Alternative, they are trying to detect different medication used, similar to marijuana, cocaine, amphetamines, and opiates.
Nevertheless, they will check for steroids,
especially in circumstances the place they’re known to be rife in a particular unit or if there is one extra reason to suspect somebody of utilizing
them.
This outcomes from providing the physique
with considerably greater amounts of male androgen hormones
like testosterone and DHT than would otherwise be produced.
When LDL cholesterol increases too much, it could put you
vulnerable to blocked arteries and raise your risk of coronary heart disease.
Eating a cholesterol-friendly food plan is crucial to reduce all risks, and
since Anavar is mainly used for chopping, your food plan isn’t likely to be a
concern. Together With plenty of wholesome
fats in the food regimen will add to danger discount.
This assumes you are not using extreme doses of
Anavar and not utilizing it continually over a protracted interval.
References:
legal steroid Uk – onlyhostess.com,
Because testosterone levels are maintained at a low stage throughout this
cycle, Anavar must serve as the primary anabolic agent.
This permits lean will increase along with the distinctive fat loss and muscle firming that happens throughout the cycle.
Utilizing Anavar throughout a steroid cycle is a highly efficient means of developing lean muscle
mass. This sturdy anabolic steroid may assist in fats
loss, muscle acquire, and efficiency enhancement. A widespread protocol for ladies is to take 50mg of
deca durabolin a week for 4-6 weeks.
Even experienced users ought to maintain vigilant monitoring and steerage.
Understanding how Oxandrolone cycles differ in terms of person experience
is integral to selling correct usage and mitigating potential complications.
A person is usually classified as a beginner or a sophisticated person, with
each class warranting distinctive cycle period and dosage considerations.
Other advantages of Anavar embody enhancing stamina
and energy as a outcome of it boosts pink blood cells.
The joint benefits of Anavar make it stand out above other slicing steroids and is considered one
of my favorite things about Anavar. Despite its status as
a highly efficient steroid, it’s nonetheless a
relatively mild oral compared with different compounds.
Anavar is by far essentially the most well-tolerated
steroid for females, in distinction to Winstrol. Oral Winstrol continues
to be thought-about a relatively mild steroid in comparison with most others.
Still, most girls will discover that Anavar is extra well-tolerated when it
comes to controlling the side effects. For women who wish to
use Winstrol, the oral type solely is recommended, and doses have to be stored very low
to keep away from virilization.
Users often utilize it to realize a well-chiseled, lean physique
and preserve muscle mass throughout extreme calorie deficits.
Anavar, significantly most well-liked by athletes and bodybuilders alike, is a famend oral anabolic steroid famous for enhancing lean muscle growth,
energy, and longevity. Its light unwanted effects and adaptability have made it a elementary element
in numerous bodily coaching routines. This would
possibly lead to intriguing questions in regards to the pace
of its effectiveness. Open the door to the transformative
impact of the anabolic steroid, generally known by its well-liked name, Anavar.
Primarily used for chopping cycles, this compound is famend worldwide
for its influence on muscle progress and energy whereas sustaining a
lean physique. Embracing this development stage involves cautious consideration, with this versatile choice gaining attention as a result of minimal undesirable outcomes and strength-enhancing
talents.
This does imply you’re using Anavar primarily as a pre-workout steroid.
In my experience, this is one of the best ways to use Anavar as a
end result of we don’t want (or even want) to maintain up constant ranges
of this liver-toxic steroid (no matter how mild) 24 hours a day.
Females working a first Anavar cycle ought to begin very low to
gauge unwanted facet effects.
The duration of Oxandrin (oxandrolone) therapy shall be decided based mostly on the patient’s response and the potential
emergence of adverse reactions. Our team of medical specialists are
there for you in every step of the greatest way, from finding the proper physician and hospital to any
sort of assistance. We educate and empower households to guarantee that
proper healthcare selections are made.
Waiting for this to naturally happen just isn’t an choice as a end result of low testosterone is significantly debilitating.
Symptoms can be widespread and critical and can embody fatigue, depression,
low libido, loss of muscle, and fat acquire, to call just a few.
Anavar is way from essentially the most suppressive steroid we can use,
however it will nonetheless have some impression in your common testosterone production32.
However being based mostly on a very powerful androgen in DHT, Anavar can include the
danger of androgenic unwanted effects in case you are somebody who is
already genetically predisposed to them. This signifies that in case you
have some male sample baldness later in life, Anavar could deliver this on earlier.
Males are much less likely to run this cycle however will want
a testosterone base (e.g., 200mg/week) and may follow with a normal Nolvadex or Clomid PCT protocol for
four to 6 weeks. Girls who are snug with larger doses of Anavar and who don’t undergo virilization results can consider including one other 5-10mg to the Anavar dose suggestion above.
This increase in muscle mass leads to an increase in metabolism,
which in turn causes an increase in vitality expenditure, resulting in a decrease in body fats.
Additionally, Anavar may help to extend muscle endurance,
which can improve athletic performance and bodily look. HGH-X2 does this
by naturally stimulating the discharge of human progress hormone with out you having to
make use of any artificial hormones. HGH-X2 is a authorized and
protected different to Somatropin, a human development hormone
(HGH) kind.
To start, Anavar suppresses anabolic steroid-related side effects like water retention and gynecomastia.
Anavar is a 17-alpha alkylated anabolic steroid (17-alkylated androgen) with the identical chemical method as oxandrolone.
This type of steroid is extraordinarily efficient at rising muscle mass while preventing
fats acquire. In this cycle Anavar dose is a bit decrease than ordinary, because of the
addition of Testosterone. So earlier than doing a stack with another steroids, first take into consideration what you
wish to obtain, after which use the correct of steroid alongside Anavar for greatest results.
For elevated effectivity, many users opt to stack Oxandrolone with compounds like testosterone,
Dianabol, and Trenbolone, relying on their fitness
objectives.
However you have to be careful of each steroids as a end result of these effects might generally result in serious health issues.
For males, the common cycle spans 6-8 weeks, while girls usually
limit it to 4-6 weeks. Prolonging the cycle past these tips could heighten the risk of unwanted side
effects, underscoring the importance of accountable and informed use.
All The Time begin with the lower finish of the dosage spectrum, especially if you’re a first-time user.
Beginning with a decrease dosage offers your body with the chance to regulate to the compound.
For beginners, 5-10 mg per day is beneficial, whereas intermediate and advanced customers can go as a lot as mg per
day. Women at our clinic have reported a gentle downturn in power, libido,
and fatigue following an Anavar cycle. With very cautious
doses (5 mg), it may be potential to avoid virilizing unwanted effects; however,
every woman might reply differently. Loopy Bulk is our beneficial source for legal steroid options, based mostly on hundreds
of optimistic reviews on verified platforms corresponding to Trustpilot and Feefo.
References:
legal anabolic steroids gnc (careers.webdschool.com)
However, it is strongly recommended to avoid alcohol consumptions at all when utilizing this steroid or some other that’s C17 alpha alkylated.
Particularly taking in consideration that the rationale
to use this steroid is the performance objective.
It could be very well known that alcohol is a direct anti performance
substance so there’s simply no sense in taking the steroid whereas consuming alcohol.
There are some things you must find out about before making a purchase.
We mean dosage rules, drug administration regimens and directions.
Elevated liver enzymes doesn’t mean that the liver is broken, nonetheless which means it may be damaged in case correct measures won’t be
taken. Although it’s hepatotoxic, it is very important notice that Oxandrolone
is one of the milder steroids as there are others out
there that are considered to be a lot far more worse. Assuming that the person is having a wholesome liver and a use that isn’t going
over the beneficial times then the liver should recover again to normal pretty quick
with out having any harm in any respect. Although it indeed can be very
helpful through the off seasons, Oxandrolone still remains
to be greatest in the course of the chopping section when the person is searching for fats burn whereas keeping the muscle tissue.
As Quickly As again, although it is not a perfect mass builder for males (but it still remains excellent for different purposes), Anavar (Oxandrolone) is an incredible steroid for girls.
As A Outcome Of we’re utilizing steroids for efficiency enhancement and bodybuilding, the compounds are being
taken at doses a lot higher than in the event that they have been used for medical purposes.
The greater your dosage, the higher your danger of adverse well being effects.
Be Taught about the benefits and dangers of steroids, how to use
them safely.
Additionally, customers are suggested to undergo liver operate checks before,
throughout, and after their cycle to observe enzyme levels.
That every little thing confirms such nice efficient and quality of this oral
steroid that among consultants in bodybuilding serves for gaining muscle mass, rising power
and improving endurance as properly. Some users
can experience hair loss from a steroid cycle, then once the cycle has completed the hair comes again thicker and fuller.
Nevertheless, if you’re taking excessive doses of Anavar or different steroids for excessive intervals of time, hair loss can turn out to be permanent.
Ladies might experience side effects such as elevated facial hair
and a deeper voice; report these symptoms to your physician instantly.
Anavar (Oxandrolone) is a synthetic steroid just like the human hormone testosterone.
The energetic ingredient on this drug is Oxandrolone, which is utilized in drugs to assist
people who are unable to gain or preserve correct weight for medical causes.
Most people asking for Anavar at present might not medically require the steroid, so doctors might
not prescribe or enable such people access to the
treatment. However, you’ll be able to still buy Anavar from reputable pharmaceutical firms.The most viable choice for getting the
medicine can be from its manufacturers. Most manufacturers ship as little
as 5 packages worldwide; they distribute the product principally
by way of their websites or retail stores.
Anavar is particularly good at getting rid of fats in more stubborn areas, and for girls, this is usually across the thighs and buttocks.
Though overall physique weight might not drop or change much, this is due
to muscle gain – fat remains to be being misplaced.
On a proper diet, ladies can count on to lose
5-10lbs of fats on an Anavar cycle unless they are lean. There’s rather more to know in regards to the unwanted effects of Anavar and all anabolic steroids, so visit my major
side effects guide here. Anavar will suppress your testosterone at just about any dosage, and the higher the dose, the extra suppression you can count on.
Its chemical composition is adjusted to exert minimal stress on the liver,
making it one of many safer decisions among oral steroids.
This lowered liver toxicity is a large alleviation for customers, particularly the ones concerned in approximately the lengthy-time period of health implications of steroid use.
This steroid is commonly used by athletes and bodybuilders to enhance
strength and performance, nevertheless it comes at a excessive value.
Due to this, powerlifters can even use Anavar to gain energy (without gaining
weight). A 20mg dose of Anavar every day may help you lose four pounds of fats
– in 12 weeks. Anabolic steroids usually scale back subcutaneous fats while increasing visceral
fats. This article will study Anavar’s benefits and the place you should purchase it.
The individual nonetheless receives a robust anabolic
impact, but the whole effect is not what he was after. Anavar has
all the time been in restricted supply ever since the first discontinuation of manufacture by Searle
within the early Nineteen Nineties. Upon resuming production a number of years later, more pharmaceutical grade merchandise have entered the market circle for many who want to buy Oxandrolone, nevertheless, pharmaceutical grade gadgets still stay pretty scarce.
Any pharmaceutical grade Anavar existing on the market is also very costly to begin with, as we will quickly see.
The safest methodology is to purchase a authorized anavar supplement such as anvarol, which mimics anavar’s results and is FDA approved.
Oxandrolone isn’t an extremely potent androgenic steroid, but
androgenic exercise does exist. Such activity can result in zits,
accelerated hair loss in these predisposed to male pattern baldness
and body hair progress. Nonetheless, most won’t have an issue
with these results as the entire androgenic nature stays very low.
An essential notice; 5-alpha reductase inhibitors are sometimes used to combat androgenic unwanted effects introduced on by way
of anabolic steroids. However, this is not going to have a robust have an result
on when utilizing Anavar because the Oxandrolone hormone
isn’t affected by the 5-alpha reductase enzyme.
Virilization signs embrace physique hair growth, a deepening of the vocal chords
and clitoral enlargement. Thankfully the virilization ranking of Anavar is extremely low; most women can complement with out falling to such effects
as lengthy as they complement responsibly. There will always be the issue of individual response to
contend with, however the Oxandrolone hormone continues to represent the safest anabolic steroid for female use.
If virilization symptoms do occur for any purpose, you’re inspired to discontinue use instantly and they’ll fade away.
Steroids, when used responsibly have been proven to accelerate muscle progress, assist in restoration, and enhance
general athletic performance.
References:
3 types of steroids (Lucille)
70918248
References:
truth about steroids (https://lauriumconsultancy.nl/companies/efectos-secundarios-de-anavar/)
Según nuestras pruebas de perfil lipídico, Winstrol es uno de los esteroides
anabólicos menos beneficiosos para el sistema cardiovascular.
Sin embargo, si los principiantes están decididos a tomar Winstrol, un ciclo de 8-10 mg/día
durante 6 semanas cut back notablemente los efectos secundarios en nuestros
pacientes. Ciertos alimentos que nos aportan vitaminas, minerales o determinadas grasas contribuyen a la síntesis de esteroides,
como la testosterona.
Hay varias formas de tomar esteroides y muchas opiniones diferentes sobre cuál es la mejor.
Algunos dependen de medicamentos como la testosterona, otros dependen de inyecciones, algunos usan pastillas o
cremas y algunos incluso optan por no usar nada. Es importante tener en cuenta
que la oxandrolona no es un sustituto de una dieta adecuada y un régimen de ejercicio.
Debe ser utilizado en combinación con un estilo de vida saludable para obtener los mejores
resultados. Para obtener los mejores resultados, debes combinar la
oxandrolona con una dieta adecuada y un programa de entrenamiento de fuerza.
Sin embargo, es importante usar este compuesto con precaución y bajo la supervisión de
un profesional de la salud para minimizar el riesgo de efectos secundarios.
Con este submit ciclo, habrá un rápido aumento de la LH,
FSH y testosterona, cubre todos los factores que podrían estar causando el retraso de llegar a su línea base
de las hormonas de forma natural. Por esta razón, creo que una vez
nuestro ciclo esté terminado es cuando deberíamos empezar este submit ciclo (una semana después de la última dosis o al día
siguiente después de tomar la última píldora . Esta regulación a la baja de los receptores de LH puede causar un aumento en el colesterol esteroidogénico (el colesterol destinado por el cuerpo para su convertirlo en testosterona).
Sin embargo, desde entonces han descubierto que la mayoría de
los beneficios de los esteroides son artificiales y pueden lograrse por medios naturales.
Ahora es común que los culturistas naturales se concentren en desarrollar músculo en lugar de aumentar el tamaño.
En la década de 2000, el culturismo evolucionó en muchas direcciones diferentes de
lo que había sido.
A pesar de lo que mucha gente piensa, el uso de esteroides no garantiza un crecimiento muscular mejor y más rápido.
Estudios importantes han encontrado que después de tomar esteroides durante un período prolongado, los efectos a menudo
disminuyen y causan efectos secundarios adversos. Los esteroides también conllevan su propio conjunto de riesgos para
la salud, como daño hepático e insuficiencia cardíaca.
Los estudios muestran que la testosterona inhibe directamente
la creación de células de grasa [7]. [newline]Como te comentaba anteriormente, estos estudios fueron realizados por científicos que sabían lo que hacían, en un ambiente controlado, con dosis controladas,
y con sustancias que SABÍAN a ciencia cierta lo que
contenían. Entre más cerca estés de tu límite genético muscular,
menos crecimiento y más lentas serán tus ganancias musculares.
Los modelos más certeros, basados científicamente, de ganancia muscular de manera natural,
predicen que los hombres pueden ganar no más de 20 a 25
libras (9.09 a 11.36 kilos) en el PRIMER AÑO de levantamiento de pesas.
La grasa corporal disminuye mientras que la hipertrofia
muscular aumenta, así como la definición muscular y la vascularidad.
El Enantato de testosterona es un éster de la hormona masculina
testosterona, ampliamente utilizado tanto en medicina como en el ámbito del culturismo.
Este compuesto se utiliza principalmente para tratar trastornos relacionados
con niveles bajos de testosterona, pero su popularidad también ha crecido en el deporte debido
a sus propiedades anabólicas. Crazy Bulk ofrece ofertas generosas, lo que permite a las mujeres ahorrar dinero, en comparación con la
compra de esteroides caros como Primobolan y Anavar en el mercado negro (que puede
costar cientos de dólares por ciclo). Este es un ciclo de corte típico utilizado
por las modelos de bikini o culturistas que desean quemar grasa.
Además del aumento en la masa muscular y la definición,
el primer ciclo de esteroides puede causar otros cambios en la apariencia física.
Algunos usuarios pueden experimentar una mayor vascularidad,
es decir, la prominencia de las venas debajo de la piel.
Esto se debe a una mayor densidad y tamaño muscular, que a su vez aumenta el flujo sanguíneo.
Otros posibles cambios podrían incluir una apariencia más “rellena” o un aumento en volumen facial debido a la retención de líquidos.
Otro aspecto visible evidente del primer ciclo de esteroides
es el aumento de la definición muscular. Los esteroides ayudan a reducir la grasa corporal, lo que permite que los músculos se vuelvan más
visibles y definidos. Esto se debe a la capacidad de los esteroides para acelerar el metabolismo y aumentar la quema de grasa,
lo que resulta en una apariencia más “cortada” y muscularmente definida.
Por otro lado, esteroides anabólicos son versiones sintéticas de la hormona macho testosterona.
Esto reduce aún más el riesgo de efectos secundarios, ya que evita una oleada repentina de testosterona exógena, manteniendo
niveles más estables en el cuerpo. Los únicos efectos secundarios reportados
por las mujeres en el estudio de one hundred
fifty mg fueron una disminución de la libido y un aumento de la fatiga, probablemente debido a una menor
producción de testosterona endógena. Sin embargo, investigaciones y pruebas reales sugieren que
el Anadrol no produce los mismos efectos androgénicos en las mujeres.
De hecho, se ha demostrado en estudios que las mujeres pueden tomar megadosis de Anadrol sin experimentar virilización. En un estudio, se administró a mujeres hasta 150 mg por día durante 30 semanas, y ninguna mostró signos de masculinización. Es importante
señalar que el riesgo de experimentar síntomas de virilización depende en gran medida
de la genética. Algunas mujeres pueden usar esteroides inadecuados durante períodos prolongados y no experimentar problemas, mientras
que otras pueden tener efectos adversos incluso
con pequeñas dosis de compuestos suaves.
Descubre cómo la Inyección de oximetolona puede potenciar tus entrenamientos y transformar tu físico de manera significativa.
Si las mujeres experimentan niveles bajos de libido o sufren de bajo bienestar,
se puede aplicar un PCT. Los efectos estimulantes del clenbuterol pueden no ser bien tolerados por mujeres sensibles
a los estimulantes, quienes pueden sentirse nerviosas, ansiosas o
experimentar insomnio. No obstante, el clenbuterol debe usarse con precaución, ya que
algunos usuarios han sido hospitalizados tras tomar dosis excesivas.
Por ejemplo, una frecuencia cardíaca de 254 latidos por minuto se registró tras una sobredosis unintentional de 10 veces la dosis recomendada.
La metandrostenolona, es un esteroide anabólico sintético derivado de la testosterona.
Por lo tanto, siempre se recomienda buscar la supervisión de profesionales de la
salud antes de considerar su uso, y seguir las pautas adecuadas
de dosificación y ciclo para minimizar los riesgos asociados.
Se ha documentado que el Dianabol puede tener efectos negativos en la salud cardiovascular, la función hepática
y los desequilibrios hormonales (7). Además de su impacto en la fuerza, el Dianabol puede
aumentar la resistencia y reducir la fatiga muscular.
La síntesis de proteínas es un proceso esencial para el crecimiento y
la reparación muscular, lo que significa que el Dianabol puede acelerar la
recuperación después del ejercicio intenso y promover el crecimiento muscular
(Kicman, 2008).
References:
Famous Baseball Players Who Used Steroids (https://Www.Dynamicviewpoint.Co.Uk/Employer/Ciclo-Clen)
70918248
References:
female bodybuilding steroids side effects (westorebd.com)
70918248
References:
testosteron Schädlich (afrikijob.com)
70918248
References:
Where can you buy legal steroids
70918248
References:
anabolic Steroids Illegal
70918248
References:
Buy steroids in the us (vijayarajastro.com)
70918248
References:
steroid pills vs injection (aryanlandmark.com)
70918248
References:
steroids and sleep – http://Www.Toycarland.com/blog.php?aidx=243818 –
70918248
References:
Legal Steroid For Muscle Building (Portalolm.Com.Br)
Заказываю каждый месяц – всегда идеально!
розы томск
70918248
References:
dbol legal steroids (Moises)
Свежесть и красота в каждом цветке!
доставка цветов томск на дом
70918248
References:
injection for muscle growth, pbs-marketing.de,
70918248
References:
what are products that Are consumed rapidly and regularly classified as?
(http://www.fabarredamenti.it)
70918248
References:
none (Louvenia)
70918248
References:
none; https://Bravepatrie.com,
70918248
References:
none – https://tourslibya.com –
Спасибо за оперативность – выручили в ДР!
доставка цветов
Доставили в больницу – всё аккуратно и бережно.
доставка цветов томск на дом
Заказывала орхидеи – нежные и красивые!
доставка цветов в томске
Качество и скорость просто на высшем уровне!
букет невесты томск
Мгновенная доставка – это просто магия какая-то!
цветы томск
Мгновенная доставка – это круто!
букет невесты томск
Учли все нюансы – виден высочайший профессионализм!
доставка цветов
Качество и скорость просто на высшем уровне!
букеты томск
Теплые слова в открытке тронули до слез!
купить пионы томск
Доставили в область – все в идеальном состоянии.
букет невесты
Муж был тронут до слез на годовщину свадьбы.
букет невесты томск
Букет простоял 10 дней – это рекорд!
букет невесты